

Abstract
The global outbreak of the COVID-19 pandemic provokes scientists to make a prompt development of new effective therapeutic interventions for the battle against SARS-CoV-2. A new series of N-(5-nitrothiazol-2-yl)-carboxamido derivatives were designed and synthesised based on the structural optimisation principle of the SARS-CoV Mpro co-crystallized WR1 inhibitor. Notably, compound 3b achieved the most promising anti-SARS-CoV-2 activity with an IC50 value of 174.7 µg/mL. On the other hand, compounds 3a, 3b, and 3c showed very promising SARS-CoV-2 Mpro inhibitory effects with IC50 values of 4.67, 5.12, and 11.90 µg/mL, respectively. Compound 3b docking score was very promising (−6.94 kcal/mol) and its binding mode was nearly similar to that of WR1. Besides, the molecular dynamics (MD) simulations of compound 3b showed its great stability inside the binding pocket until around 40 ns. Finally, a very promising SAR was concluded to help to design more powerful SARS-CoV-2 Mpro inhibitors shortly.
Graphical Abstract
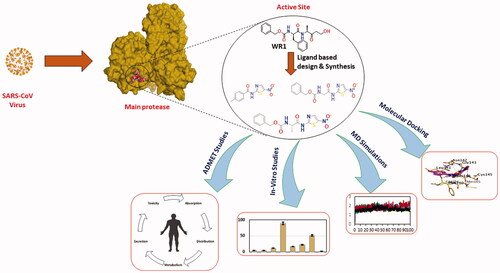
A new series of N-(5-nitrothiazol-2-yl)-carboxamido derivatives were designed and synthesised based on the structural optimisation principle.
In vitro antiviral activities against SARS-CoV-2 using SARS-CoV-2 cell-based inhibitory assay.
The anticipated inhibitory effects of the synthesised compounds (3a–g) towards the SARS-CoV-2 Mpro enzyme were emphasised by using the SARS-CoV-2 Mpro assay.
Molecular docking studies, molecular dynamics simulations for 100 ns, and MM-GBSA calculations were carried out for the newly synthesised compounds (3a–g) compared to the co-crystallized inhibitor (WR1).
ADMET and toxicity in silico studies were applied for the designed derivatives.
Finally, our interesting work rationale helped to conclude a very promising structure-activity relationship (SAR) finding.
HIGHLIGHTS
Acknowledgements
The authors would like to extend their sincere appreciation to Taif University ResearchSupporting project number (TURSP-2020/330). Eman Y. Santali would like to thank TaifUniversity, Taif, Saudi Arabia, for providing computer facilities for running MD simulations.Mohamed Elagawany would like to thank the Science and Technology Development Fund(Egypt) for financial support (STDF-TDG Grant 43324).
Author contributions
Mohamed Elagawany and Ahmed A. Al-Karmalawy: conceptualization. Mohamed Elagawany, Ayman Abo Elmaaty, Bahaa Elgendy, and Ahmed A. Al-Karmalawy: formal Analysis. Mohamed Elagawany, Eman Y. Santali, and Ahmed A. Al-Karmalawy: funding acquisition. Ayman Abo Elmaaty, Ahmed Mostafa, Noura M. Abo Shama, Eman Y. Santali, and Ahmed A. Al-Karmalawy: methodology. Mohamed Elagawany and Ahmed A. Al-Karmalawy: project administration and resources. Ayman Abo Elmaaty and Ahmed A. Al-Karmalawy: software. Ahmed A. Al-Karmalawy: supervision. Ayman Abo Elmaaty and Ahmed A. Al-Karmalawy: validation. Mohamed Elagawany, Ayman Abo Elmaaty, Ahmed Mostafa, Eman Y. Santali, Bahaa Elgendy, and Ahmed A. Al-Karmalawy: writing—original draft. Mohamed Elagawany, Ayman Abo Elmaaty, Ahmed Mostafa, and Ahmed A. Al-Karmalawy: writing—review and editing. All authors approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).