
Figures & data
Table 1. Mutation or overexpression of JAK/STAT at different lymphoma diseases.
Figure 1. (A) Activation and regulation of the canonical JAK/STAT signalling pathway: cytokine binding to its receptor recruits and activates JAKs coupled to the receptor, which phosphorylates specific tyrosines on the bound receptor and creates docking sites for proteins with the SH2 structural domain (including STATs). Subsequently, JAKs mediate the phosphorylation of STATs to form homodimers/heterodimers into the nucleus, which bind to the promoters of the corresponding target genes to activate the transcription and expression of the relevant genes. (B) Abnormal activation of the JAK/STAT pathway in lymphoma: 1. In HL and PMBCL, the abnormal amplification of the 9p24.1 gene in the chromosome of tumour cells leads to an increase in the expression of PD-L1 and JAK2 at this position, and the continuous activation of the JAK/STAT signalling pathway further promotes the increase in the expression of PD-L1, and the high-expression of PD-L1 binds to PD-1 on the surface of the T cells, causing the T cells to ‘over-deplete’ and loss function of T cells, leading to the escape of tumour cells from immune surveillance. 2. In CTCL, JAK3 mutations lead to an increase in activity and an abnormal activation of the downstream STAT5, while a variety of cytokines secreted by the tumour cells also lead to the upregulation of the constitutive activation of the STAT5, leading to the survival and development of malignant tumour cells.3. In HL, STAT3, STAT5, STAT6 can be abnormally activated and expressed at high levels, accelerating tumour development.
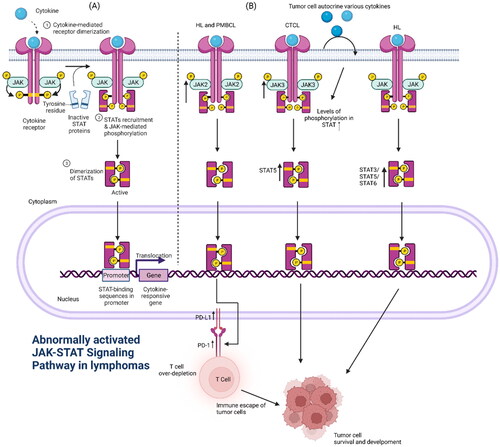
Figure 2. Type I PI3K is composed of the regulatory subunit P85 and the catalytic subunit P110. It can be divided into two subtypes, IA and IB, which are activated in two major ways: (1) interaction of PI3K with growth factor receptors or ligand proteins that have tyrosine residues and are phosphorylated (2) direct binding of p110 through Ras. Activated PI3K activates AKT by phosphorylating its substrate PIP2 to PIP3. In addition, it activates the mTOR protein through phosphorylation. mTOR includes two polymolecular complexes, namely, mTORC1 and mTORC2, which regulate protein synthesis and cell growth. Abnormal activation of the PI3K/AKT/mTOR pathway in lymphoma: 1. Mutations in the PIK3CA gene encoding p110α in patients with DLBCL and increased copy number of PIK3CA in patients with MCL lead to aberrant activation of the PI3K/AKT/mTOR pathway.2 In some patients with lymphoma, the phosphorylation of AKT is markedly increased, which leads to the inhibition of the function of the activated AKT on the cell-cycle inhibitors P21 and P27, resulting in uncontrolled tumour cells growth.3 High expression of AKT directly phosphorylates downstream FOX03a, which is associated with poor prognosis in patients with MCL.3 PTEN function, which is opposite to PIK3, inhibits AKT function, and inhibits downstream FOX03a, which is associated with poor prognosis in patients with MCL. The high expression of AKT directly phosphorylates downstream FOX03a, which is significantly associated with poor prognosis in MCL patients.3. PTEN functions in the opposite way to PIK3, inhibiting AKT activation and blocking signalling, and the lack of PTEN in GCB-type DLBCL patients leads to aberrant activation of the PI3K/AKT/mTOR signalling pathway.
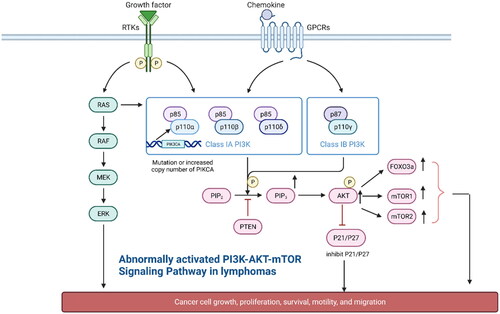
Table 2. Distinct activation of the PI3K-AKT-mTOR pathway at different lymphoma diseases.
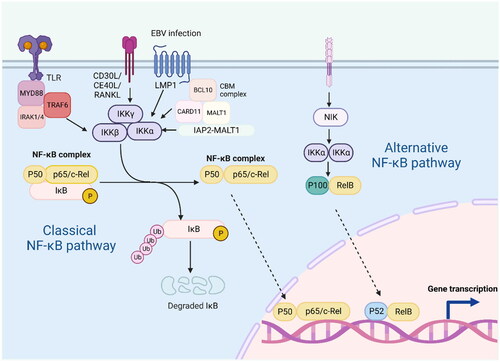
Figure 3. The classical and alternative pathways of NF-KB signalling are mediated by a variety of receptors and signalling molecules, including TLR, CD30L, CD40L, RANKL, etc.; persistent EBV infection can also activate this pathway. In the classical NF-KB pathway, IKB binds to the P50-P65 heterodimer in the cytoplasm, preventing its entry into the nucleus. When receptor activation initiates adaptor proteins and signalling kinase responses, this will lead to activation of the IKB kinase complex(IKK), which phosphorylates IKB and leads to polyubiquitination and proteasomal degradation. This allows P50-P65 to be released into the nucleus to induce gene expression. In the alternative pathway, IKKα is activated by the upstream kinase NF-KB inducible kinase (NIK), which promotes the processing of P100-RelB into P52-RelB. Abnormal activation of the NF-KB pathway in lymphoma: 1. In MALT, CARD11, MALT1 and BCL10 are involved in the formation of the complex CBM, which is essential for the activation of NF-KB. 2. Meanwhile, overexpression of IAP2-MALT1 fusion proteins in MALT is also effective in activating NF-KB.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.