Figures & data
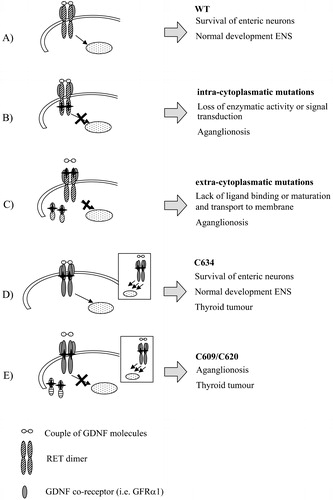
Figure 1. Distribution ofRET mutations along the 21 exons. Missense, nonsense and deletion/insertion mutations conferring loss of function, together with gain‐of‐function and splicing mutations are reported using different symbols. The predisposing haplotype and relative position of its specific markers are indicated by a striped rectangle. Protein domains are reported as well. SP = signal peptide; CDL = cadherin‐like domain; CR = cysteine‐rich domain; TM = transmembrane; TK = tyrosine kinase domain.
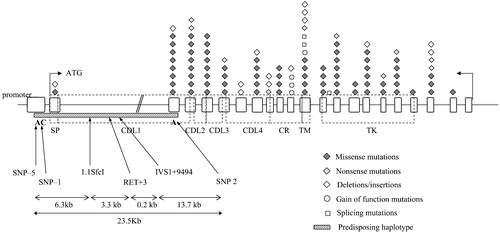
Figure 2. Pathogenetic mechanism of different RET mutations in enteric nervous system(ENS) development. The RET signalling complex is formed by a RET dimer, the ligand GDNF and co‐receptor GRFα1 (A). Intracytoplasmatic RET mutations (B) can lead to loss of activity of the RET protein, while in presence of extracellular mutations (C) either lack of binding activity or lack of exposure to the cell surface due to misfolding can occur. Mutations of cysteine residues of the cysteine‐rich domain confer a gain of function in thyroid tissues (represented in the boxes) leading to tumour. Depending on cysteine codons involved in the substitutions, either normal (D) or impaired (E) ENS development is achieved.