Figures & data
Figure 1. General aspects of RNOS in aging. Young individuals have a higher stress tolerance due to a high antioxidant potential, and efficient repair mechanisms restore homeostatic conditions in response to damage. Under these physiological conditions redox regulation can normally proceed in special compartments but differs in a cell type-specific manner. Prostaglandin endoperoxide H2 synthase (PGHS, COX) is an excellent example of a highly redox-controlled enzyme. In aging the stress tolerance to RNOS decreases. The molecular basis for the loss of balance between damage and repair is the enhanced RNOS generation which must be compensated by antioxidant systems. These exhibit a reduced efficiency in repair with increasing age. As a consequence, oxidatively altered macromolecules accumulate and burden physiological cellular processes. This attenuates redox regulation and also interferes with intercellular communication.
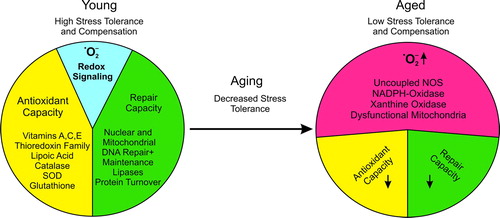
Table I. Theories of aging.
Table II. Major cellular NO sources and regulation.
Figure 2. The vascular •NO signaling cascade and the antagonizing •O2−. Agonist stimulation of endothelial cells, including angiotensin II, acetylcholine, serotonin, etc., or shear stress causes activation of endothelial NO synthase by a transient increase in intracellular calcium or phosphorylation. •NO as a freely diffusible molecule can easily cross cell membranes and activate soluble guanylyl cyclase in smooth muscle cells, increasing intracellular cGMP levels. Protein kinase G is activated and can cause a decrease in intracellular calcium [Ca2 + ]i and mediates the dephosphorylation of myosin light chain leading to vasorelaxation. Superoxide can react with and reduce free •NO, impairing the •NO-cGMP signaling pathway. Inactivation of soluble guanylyl cyclase by superoxide has been reported, too. Various superoxide sources in the endothelium and vascular smooth muscle cells have been identified, including mitochondria, uncoupled NO synthase, xanthine oxidase, NADPH oxidases, and epoxygenase. Depending on the model and pathology, different sources of •O2− can be activated. (Ag = agonist; [Ca2 + ]i = intracellular calcium; NOS-3 = endothelial NO synthase; sGC = soluble guanylyl cyclase; •NO = nitric oxide; cGMP = cyclic guanosine monophosphate; Rec = receptor; •O2− = superoxide anion)
![Figure 2. The vascular •NO signaling cascade and the antagonizing •O2−. Agonist stimulation of endothelial cells, including angiotensin II, acetylcholine, serotonin, etc., or shear stress causes activation of endothelial NO synthase by a transient increase in intracellular calcium or phosphorylation. •NO as a freely diffusible molecule can easily cross cell membranes and activate soluble guanylyl cyclase in smooth muscle cells, increasing intracellular cGMP levels. Protein kinase G is activated and can cause a decrease in intracellular calcium [Ca2 + ]i and mediates the dephosphorylation of myosin light chain leading to vasorelaxation. Superoxide can react with and reduce free •NO, impairing the •NO-cGMP signaling pathway. Inactivation of soluble guanylyl cyclase by superoxide has been reported, too. Various superoxide sources in the endothelium and vascular smooth muscle cells have been identified, including mitochondria, uncoupled NO synthase, xanthine oxidase, NADPH oxidases, and epoxygenase. Depending on the model and pathology, different sources of •O2− can be activated. (Ag = agonist; [Ca2 + ]i = intracellular calcium; NOS-3 = endothelial NO synthase; sGC = soluble guanylyl cyclase; •NO = nitric oxide; cGMP = cyclic guanosine monophosphate; Rec = receptor; •O2− = superoxide anion)](/cms/asset/0353c7af-ae72-4238-9ed6-364f03e10686/iann_a_645498_f0002_b.jpg)
Table III. Potential sources of superoxide in the vasculature.
Figure 3. The Yin–Yang system of prostanoids. Agonist stimulation of endothelial cells causes an increase in intracellular calcium levels [Ca2 + ]i, which releases arachidonic acid via activation of phospholipase A2 from membrane phospholipids. Alternatively, phospholipase A2 is regulated by fluid or cyclic shear stress through phosphorylation. Arachidonic acid is converted by prostaglandin endoperoxide H2 synthase (cyclo-oxygenase) into the intermediate prostaglandin endoperoxide H2, which is further processed into prostacyclin by prostacyclin synthase. Prostacyclin mediates vasorelaxation by acting on the prostacyclin receptor, activating adenylyl cyclase, and elevating cyclic AMP, resulting in an increase in protein kinase A activity. Furthermore, prostacyclin inhibits platelet aggregation and leukocyte adhesion to the endothelium. Under conditions that cause inhibition of prostacyclin synthase, PGH2 levels increase and PGH2 exerts thromboxane-like effects via stimulation of the thromboxane A2/PGH2 receptor. Thromboxane A2 stimulates platelet aggregation and enhances this process in an autocrine loop or promotes leukocyte adhesion. Activation of the thromboxane A2/PGH2 receptor leads to activation of phospholipase C and an increase in intracellular calcium levels. (AA = arachidonic acid; AC = adenylyl cyclase; Ag = agonist; [Ca2 + ]i = intracellular calcium; G = G-protein; ONOO− = peroxynitrite; •O2− = superoxide; PLA2 = phospholipase A2; •NO = nitric oxide; PGHS-1 = prostaglandin endoperoxide H2 synthase; PGH2 = prostaglandin endoperoxide H2; PGIS = prostacyclin synthase; PLC = phospholipase C; PKA = protein kinase A; Rec = receptor; TxA2 = thromboxane A2; TxAS = thromboxane synthase)
![Figure 3. The Yin–Yang system of prostanoids. Agonist stimulation of endothelial cells causes an increase in intracellular calcium levels [Ca2 + ]i, which releases arachidonic acid via activation of phospholipase A2 from membrane phospholipids. Alternatively, phospholipase A2 is regulated by fluid or cyclic shear stress through phosphorylation. Arachidonic acid is converted by prostaglandin endoperoxide H2 synthase (cyclo-oxygenase) into the intermediate prostaglandin endoperoxide H2, which is further processed into prostacyclin by prostacyclin synthase. Prostacyclin mediates vasorelaxation by acting on the prostacyclin receptor, activating adenylyl cyclase, and elevating cyclic AMP, resulting in an increase in protein kinase A activity. Furthermore, prostacyclin inhibits platelet aggregation and leukocyte adhesion to the endothelium. Under conditions that cause inhibition of prostacyclin synthase, PGH2 levels increase and PGH2 exerts thromboxane-like effects via stimulation of the thromboxane A2/PGH2 receptor. Thromboxane A2 stimulates platelet aggregation and enhances this process in an autocrine loop or promotes leukocyte adhesion. Activation of the thromboxane A2/PGH2 receptor leads to activation of phospholipase C and an increase in intracellular calcium levels. (AA = arachidonic acid; AC = adenylyl cyclase; Ag = agonist; [Ca2 + ]i = intracellular calcium; G = G-protein; ONOO− = peroxynitrite; •O2− = superoxide; PLA2 = phospholipase A2; •NO = nitric oxide; PGHS-1 = prostaglandin endoperoxide H2 synthase; PGH2 = prostaglandin endoperoxide H2; PGIS = prostacyclin synthase; PLC = phospholipase C; PKA = protein kinase A; Rec = receptor; TxA2 = thromboxane A2; TxAS = thromboxane synthase)](/cms/asset/6f0509ab-6a04-4aef-b2cc-e32afb486d3f/iann_a_645498_f0003_b.jpg)
Figure 4. Free radical-mediated platelet hyperreactivity in aging. Platelets use peroxynitrite at low concentrations (< 21 nM) in order to provide the peroxide tone for modulating basal prostaglandin endoperoxide H2 synthase (cyclo-oxygenase-1) activity after agonist-mediated platelet activation. NADPH oxidase and endothelial NO synthase concertedly release superoxide anion and •NO to form peroxynitrite. Cyclo-oxygenase-1 provides the substrate prostaglandin endoperoxide H2 for thromboxane synthase, which is converted into thromboxane A2. This potent eicosanoid further activates platelets in an autocrine loop or causes the recruitment of other platelets to the thrombus. However, aging leads to an elevated superoxide formation via uncoupling of NO synthase, conversion of xanthine dehydrogenase into an oxidase, activation of NADPH oxidase, or by electron leakage in the mitochondrial electron transport chain. This saturates the platelet peroxide tone, resulting in loss of the modulation of COX-1 activity, which subsequently leads to hyperreactivity of platelets. Therefore, platelet aggregation readily occurs at lower doses of agonists and proceeds faster. (Ag = agonist; Rec = receptor; PLA2 = phospholipase A2; PGHS-1 = prostaglandin endoperoxide H2 synthase; AA = arachidonic acid; PGH2 = prostaglandin endoperoxide H2; TxAS = thromboxane synthase; ONOO− = peroxynitrite; TxA2 = thromboxane A2; [Ca2 + ]i = intracellular calcium; •NO = nitric oxide; •O2− = superoxide)
![Figure 4. Free radical-mediated platelet hyperreactivity in aging. Platelets use peroxynitrite at low concentrations (< 21 nM) in order to provide the peroxide tone for modulating basal prostaglandin endoperoxide H2 synthase (cyclo-oxygenase-1) activity after agonist-mediated platelet activation. NADPH oxidase and endothelial NO synthase concertedly release superoxide anion and •NO to form peroxynitrite. Cyclo-oxygenase-1 provides the substrate prostaglandin endoperoxide H2 for thromboxane synthase, which is converted into thromboxane A2. This potent eicosanoid further activates platelets in an autocrine loop or causes the recruitment of other platelets to the thrombus. However, aging leads to an elevated superoxide formation via uncoupling of NO synthase, conversion of xanthine dehydrogenase into an oxidase, activation of NADPH oxidase, or by electron leakage in the mitochondrial electron transport chain. This saturates the platelet peroxide tone, resulting in loss of the modulation of COX-1 activity, which subsequently leads to hyperreactivity of platelets. Therefore, platelet aggregation readily occurs at lower doses of agonists and proceeds faster. (Ag = agonist; Rec = receptor; PLA2 = phospholipase A2; PGHS-1 = prostaglandin endoperoxide H2 synthase; AA = arachidonic acid; PGH2 = prostaglandin endoperoxide H2; TxAS = thromboxane synthase; ONOO− = peroxynitrite; TxA2 = thromboxane A2; [Ca2 + ]i = intracellular calcium; •NO = nitric oxide; •O2− = superoxide)](/cms/asset/6356a6ff-cf80-4706-83ff-7ac12af05858/iann_a_645498_f0004_b.jpg)