
Figures & data
Figure 1. The relationship between melatonin and endothelial dysfunction in hepatic IRI. Melatonin exerts hepatoprotective effects via inhibition of iNOS, stimulation of eNOS, and reduction of TNF-α and ET-1. Endothelial dysfunction leads to vascular stress and remodeling and to the expression of pro-inflammatory and pro-apoptotic genes, thus worsening hepatic IRI. eNOS = endothelial NO synthase; ET-1 = endothelin-1; iNOS = inducible NO synthase; IRI = ischemia/reperfusion injury; TNF-α = tumor necrosis factor α.
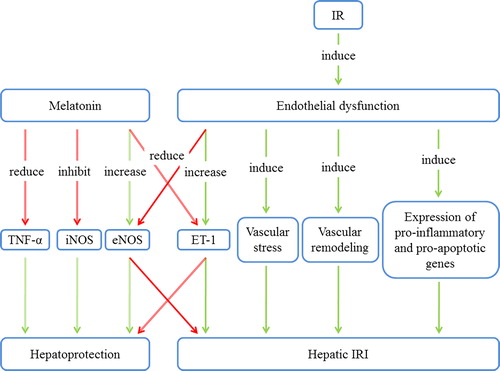
Figure 2. The involvement of mitochondria in apoptotic cell death and cellular necrosis. The overproduction of ROS by the mitochondria causes lipid peroxidation. Lipid peroxidation increases the permeability of the mitochondrial membrane. The increased membrane permeability ultimately leads to a loss of mitochondrial integrity that results in the release of cytochrome c into the cytoplasm, the subsequent activation of caspase activity, and the initiation of apoptotic cell death. ROS = reactive oxygen species.
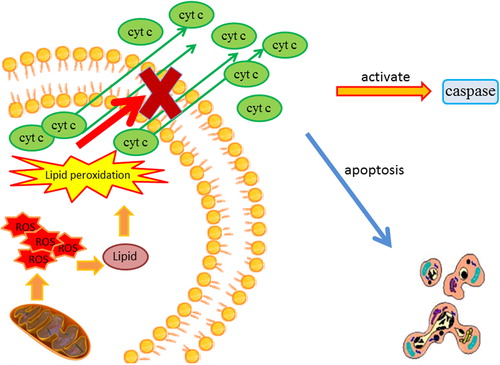
Table I. The effects of melatonin on experimental models subjected to hepatic IRI.
Figure 3. The downstream pathways of melatonin in hepatic IRI. Melatonin increases HO-1 and inhibits JNK. HO-1 inhibits JAK2/STAT1, TRIF, IFN-β, CXCL-10, and MyD88. Bicyclol, NAC, neutrophils, and repressed MyD88 cause attenuation of TLR4. TLR4 repression leads to the suppression of NF-κB. These changes result in hepatoprotection. CXCL-10 = C-X-C motif ligand 10; HO-1 = heme oxygenase 1; IRI = ischemia/reperfusion injury; JAK2 = Janus kinase 2; JNK = c-Jun N-terminal kinase; MyD88 = myeloid differentiation factor 88; NAC = N-acetylcysteine; NF-κB = nuclear factor κB; STAT1 = signal transducer and activator of transcription 1; TLR-4 = toll-like receptor 4; TRIF = toll-receptor-associated activator of interferon.
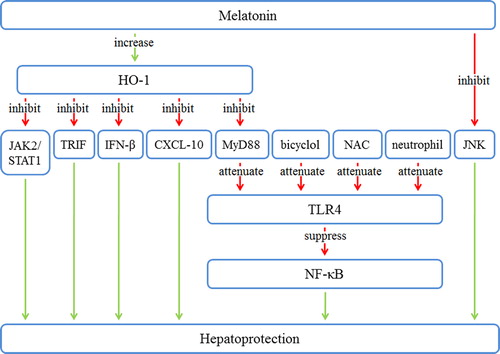
Table II. Clinical evidence supporting the protective effects of melatonin in liver diseases.