Figures & data
Figure 1. The role of oxidative stress in diabetes pathogenesis. Decreased antioxidant defense, hyperglycemia, inflammation, and obesity contribute to the ROS increase in the body. Elevated levels of ROS can damage carbohydrates, amino acids, and lipids leading to the formation of RCC. RCC can further react with macromolecules yielding advanced glycation or lipoxidation end products, AGE or ALE, respectively. Both AGE and ALE have important role in the onset and progression of diabetes.
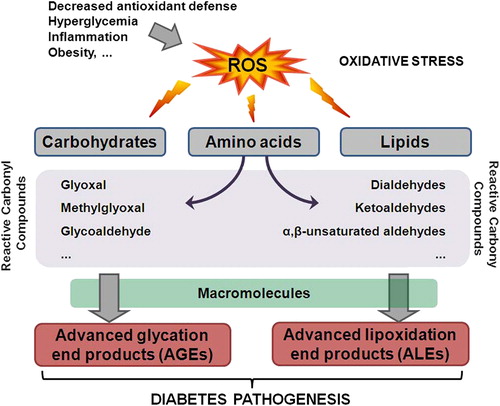
Figure 2. Oxidative stress and HNE impair insulin signaling. Hyperglycemia and hyperlipidemia induce ROS generation, which in turn promote lipid peroxidation and formation of reactive aldehydes (e.g. HNE) that can further bind to IRS and impair IRS function. Elevated intracellular ROS can damage DNA and lead to the formation of 8OHG. In addition, ROS can induce UCP2 in mitochondria or inactivate PDX1. Furthermore, activation of IRS1 leads to phosphorylation of FOXO1 transcription factor and FOXO1 nuclear export. However, ROS can either directly induce acetylation of FOXO1 or through JNK phosphorylate FOXO1 at different residues and thus promote FOXO1 nuclear translocation. FOXO1 also competes for the PDX1 promoter and represses the expression of PDX1 that is responsible for β-cell proliferation.
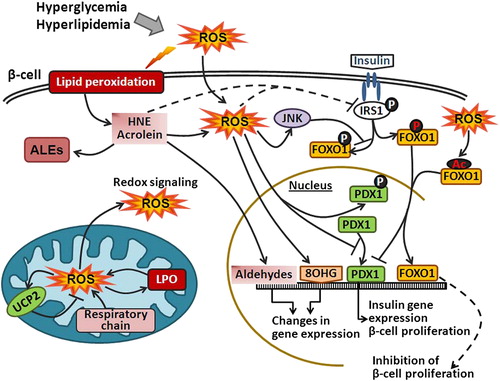
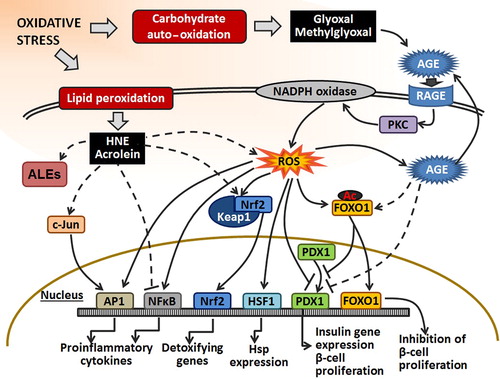
Figure 3. The effect of RCC on transcription factors. Under oxidative stress, RCC are formed by auto-oxidation of carbohydrates (e.g., glyoxal and methylglyoxal) or by lipid peroxidation (e.g., HNE and acrolein). LPO-derived RCC can react with NFκB and suppress its action. Also, they can indirectly activate AP1 and Nrf2 by phosphorylation of c-Jun or alkylation of Keap1 in the Keap1–Nrf2 complex, respectively. Moreover, they can affect transcription factors indirectly through ROS signal transduction pathways. The effect of RCC, derived from carbohydrate oxidation, on the transcription factors is either achieved through AGE or ROS transduction pathway. AGE can promote acetylation of FOXO1 that can inhibit proliferation of β-cells and insulin gene expression. Also, binding of AGE to AGE receptor (RAGE) stimulates ROS production by PKC and NADPH oxidase. Furthermore, elevated ROS can activate AP1, NFκB, and HSF1, and inhibit PDX1 binding to DNA. Accordingly, they induce pro-inflammatory gene expression, Hsp expression, and inhibit insulin gene expression and β-cell proliferation. Also, ROS can induce dissociation of Nrf2 from Keap1–Nrf2 complex enabling Nrf2 translocation to the nucleus and activation of the expression of detoxifying genes.