Figures & data
Figure 1. DRAs and related nucleic acid-dependent ATPases. (A) The conserved SF2 ATPase/helicase core. The figure is prepared from eIF4aIII, which is a component of the exon junction complex (PDB: 2J0S). Positions of the characteristic motifs are highlighted. (B) Domain organization of DRAs. Domains are not to the scale. C-terminal regions that resemble the ATPase domains of Dicer and FANCM-like proteins are simplified and are not labeled. (C) Schematic cladogram showing DRAs within the SF2 family of proteins, specifically the DEAD box family and double stranded nucleic acid binding ATPases. The alignment and family trees were determined with the UGENE software package.Citation118 The multiple sequence alignment was run with T-CoffeeCitation119 on the core ATPase/helicase domains listed in and the family tree was determined using the PHYLIP Neighbor Joining method with the Jones-Taylor-Thornton distance matrix. Pair-wise sequence identity for the ATPase core regions of DRAs range from the highest 42% (hsMDA5: hsLGP2) and 36% (hsRIG-I: hsMDA5), to 26% (hsRIG-I: ceDRH-1) and 21% (ceDRH-1: ceDRH-3), with the lowest 14% between hsDicer1 and ceDRH-3.
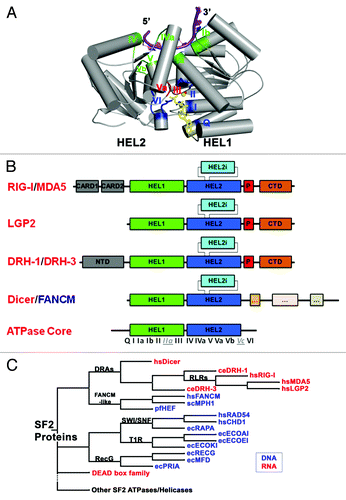
Table 1. SF2 nucleic acid-dependent ATPases listed in this study
Figure 2. Sequence and structural features of RIG-I that contribute to duplex RNA recognition. (A) Sequence alignment of DRAs and other SF2 proteins. Notice motifs IIa and Vc (boxed in dotted lines) are not very conserved in amino acid sequence. (B) HEL2i domain juxtaposes with the duplex RNA backbone (PDB codes: 3TMI in green; 2YKG in yellow; 4A36, in magenta). (C) Specialized motifs IIa and Vc recognize the top strand of the duplex RNA (Botton strand or tracking strand is the strand nucleic acid that binds to the SF2α proteins; Top strand is the complementary strand). (D) Possible structural conservation of motif IIa and motif Vc found in DEAD-box RNA family members. Figure shows the aligned structures of DEAD-box protein:ssRNA complexes with duplex RNA (PDB codes: 2J0S, 3I5X, 3G0H and 2DB3). The possible presence of motif IIa and Vc in DEAD-box proteins are labeled in parenthesis.
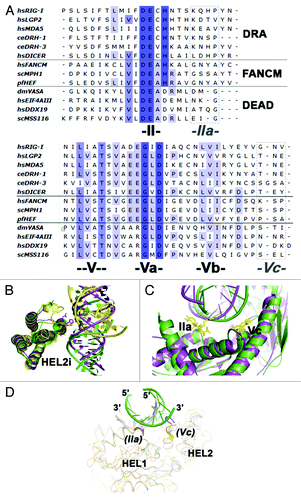
Figure 3. (A) Segmented map of human Dicer with crystal structures of homologous domains docked. (B) Model for pre-miRNA recognition. A pre-miRNA hairpin is modeled into the proposed binding channel of Dicer, with the stem-loop fit in the RNA-binding cleft of the protein. (C) Schematic for processive dicing in which dsRNA is translocated into the nuclease core (1). The PAZ domain (purple) recognizes the dsRNA end, positioning RNase III (orange) for cleavage (2). The siRNA product is released while the dsRNA substrate remains bound to the protein (3). Reprinted with permission from Lau et al.Citation44
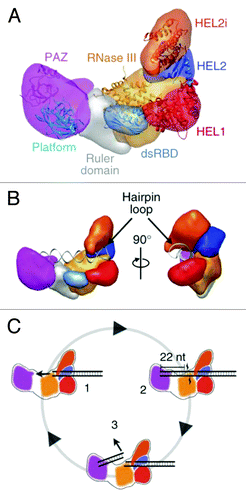
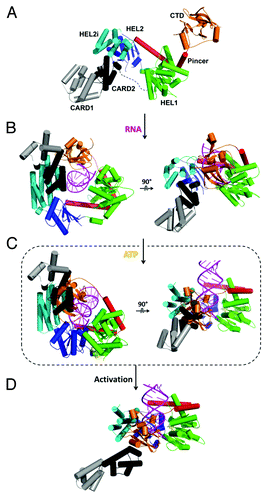
Figure 4. Structural basis for dsRNA recognition and activation of RLRs. Models were created by aligning and merging known duck and human RIG-I structures and by taking consideration of our recent solution hydrodynamic studies on RIG-I conformational dynamics upon RNA and ATP binding.Citation36 (A) Model of full length RIG-I apoenzyme based on structures of duck RIG-I (PDB: 4A2W) and the CTD (PDB: 4A2V). In the autoinhibited conformation, the N-terminal CARDs are sequestered from signaling and maintain RIG-I in an autoinhibited state. (B) RIG-I switches into a semi-closed conformation upon RNA binding. Binding of dsRNA to the CTD brings the HEL domains in contact with dsRNA (PDB: 4A2Wand 2YKG). (C) ATP binding (yellow) closes the HEL domains that would cause a clash between the CARDs and CTD (PDB: 4A2W, 4A36 and 2YKG). (D) The change in conformation upon dsRNA and ATP binding releases the CARD domains for signaling.