
Abstract
Objective
Among patients with enteropancreatic neuroendocrine tumor syndromes only one case with a cholecystokinin (CCK) secreting tumor has been reported. She had significant hyperCCKemia leading to a specific syndrome of severe diarrheas, weight loss, repeated duodenal ulcers and a permanently contracted gallbladder with gallstones. There are, however, reasons to believe that further CCKomas exist, for instance among Zollinger–Ellison patients with normal plasma gastrin concentrations. The present review is a call to gastroenterologists for awareness of such CCKoma patients.
Method
After a short case report, the normal endocrine and oncological biology of CCK is described. Subsequently, the CCKoma symptoms are discussed with particular reference to the partly overlapping symptoms of the Zollinger-Ellison syndrome. In this context, the diagnostic use of truly specific CCK and gastrin assays are emphasized. The discussion also entails the problem of access to accurate CCK measurements.
Conclusion
Obviously, the clinical awareness about the CCKoma syndrome is limited. Moreover, it is also likely that the knowledge about the necessary specificity demands of diagnostic gastrin and CCK assays have obscured proper diagnosis of the CCKoma syndromes in man.
Introduction
Endocrine tumors in the gut and the pancreas are diagnosed with increasing frequency [Citation1,Citation2]. Some of the tumors secrete hormones in amounts that cause clinical syndromes with a symptomatology characteristic for the expressed gut or pancreas hormone. Well-known examples are the syndromes of gastrinomas, glucagonomas, insulinomas, somatostatinomas and VIPomas (tumors expressing vasoactive intestinal polypeptide) (for review, see ref. Citation3).
Cholecystokinin (CCK) is – as recently detailed [Citation4] – a classical gut hormone, which, however, is widely expressed also in extraintestinal tissues. Thus, CCK has been shown to be expressed in particularly high concentrations in cerebral and peripheral neurons [Citation5–7]. In modest amounts, however, CCK is also expressed in endocrine neoplasias such as the pituitary corticotrophic Cushing and Nelson tumors [Citation8], in medullary thyroid carcinomas [Citation9], in pheochromocytomas [Citation10] as well as in nonendocrine tumors such as Ewing sarcomas [Citation11], some gliomas [Citation12], and acoustic neuromas [Citation13]. Notably, increased plasma concentration of bioactive CCK peptides with ensuing symptoms in patients with the above-mentioned neuroendocrine tumors has so far not been reported. However, a metastatic cell line of a rat islet-cell tumor expressed CCK in significant amounts, which led to lethal anorexia and cachexia after transplantation into mice [Citation14,Citation15]. Moreover, we recently found a patient with excessive CCK release from a pancreatic islet-cell tumor and its liver metastases [Citation16,Citation17]. Her symptoms are fully explained by high concentrations of bioactive CCK peptides in plasma. Thus, her case constitutes, to the best of our knowledge, the first reported hyper-CCKemic tumor syndrome in man, a CCKoma syndrome [Citation17].
It is a general experience with endocrine tumor syndromes that the clinical phenotype may vary individually, partly related to the concentration of the hormone in plasma. It is also well known that tumor syndromes may illustrate essential aspects of function, biochemistry and pathophysiology of a given hormone, as has been shown by gastrinomas [Citation18,Citation19]. Therefore, it is important that further cases of new and perhaps rare syndromes are uncovered and reported in detail – both for the sake of the individual patients suffering from a new syndrome and for the general knowledge about endocrine tumors. The CCKoma syndrome is no exception in these respects. In spite of the detailed report published eight years ago [Citation17] about the singular Danish CCKoma patient, we are, however, lacking descriptions of additional CCKoma patients. Hopefully, however, this review will stimulate interest in uncovering further cases and awareness of the diagnostic challenges among patients with enteropancreatic endocrine tumors.
A short case report
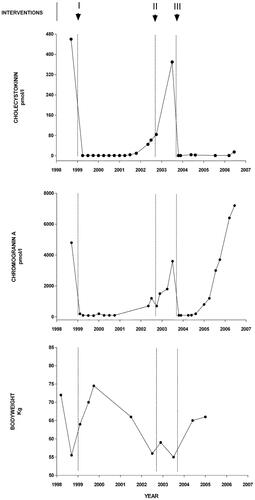
A 58-year-old woman was admitted for examination of a neuroendocrine tumor. For one year she had had frequent episodes of diarrhea and a weight loss of 15 kg, but no abdominal pain or flushing. She also had dyspeptic symptoms and was treated with proton-pump inhibitors after an episode of hematemesis. Ultrasound scanning at a regional hospital revealed processes in the liver, biopsies of which contained chromogranin A and synaptophysin-positive cells consistent with a neuroendocrine tumor. The primary tumor had not been located, but treatment with octreotide and streptozotocin plus fluorouracil was initiated with beneficial effect. The diarrhea stopped and the patient regained weight and plasma chromogranin A decreased to normal concentrations (). In a new biopsy of the liver metastases, the tumor cells again stained positive for chromogranin A and synaptophysin, but neither insulin, glucagon, somatostatin, VIP, nor serotonin could be demonstrated. Later, the weight had returned to normal (75 kg) and the patient was generally well. After two years, however, the diarrheas reappeared. The weight decreased to 56 kg, the chromogranin A concentration in plasma rose and the patient had recurrent bilestone attacks. Ultrasound scanning showed multiple small stones in a contracted gallbladder. Later, a large bleeding ulcer in the duodenum was found. It was successfully treated with a proton-pump inhibitor. Gastrin concentrations in plasma had been measured regularly and were always within the normal reference range. Octreotide scintigraphy then revealed two large metastases in the right lobe of the liver and a tumor in the tail of the pancreas. Subsequent distal pancreatectomy revealed a tumor of 5 cm in diameter that contained chromogranin A. Neither insulin, glucagon, gastrin, nor serotonin positive cells were found. The Ki67 proliferation index was then 5%. Biopsies of the liver metastases showed only fibrosis and inflammation. Resection of the primary tumor in the pancreas, however, had no effect on the diarrhea, but only caused a minor decrease in the chromogranin A concentrations (). A few months later, the chromogranin A concentrations rose, and right-sided hemihepatectomy with cholecystectomy were performed. At that time, metastases were found only in the right liver lobe. Immunostaining confirmed the neuroendocrine phenotype but revealed no insulin, glucagon, gastrin, somatostatin, serotonin, or neuron-specific enolase (NSE). The metastatic Ki67 proliferation index was now 30%. Following hemihepatectomy, the diarrhea stopped and chromogranin A in plasma decreased to normal for six months. Later, however, the chromogranin A concentrations again increased (). Recurrence of multiple metastases in the remaining liver was observed, but further chemotherapy or radionuclide treatment was not possible due to severely decreased kidney function, and the patient died from disseminated disease. A detailed case report has been published in ref. [Citation17].
Figure 1. Cholecystokinin (CCK) and chromogranin A concentrations in plasma related to bodyweight in the CCKoma patient: the upper panel shows the variation in CCK concentrations from admission (december 1998) to death (august 2006). The patient was fasting at all blood samplings, even though the meal variation in plasma CCK in healthy subjects is limited (<1 to 5 pmol/l). The initial chemotherapy (I) with Octreotide-LAR and streptozotocin rapidly normalized the plasma CCK and chromogranin A (mid panel) concentrations with an effect lasting for two and a half years. In late 2001 and in 2002, the concentrations rose again. Surgical removal of the neuroendocrine tumor in the pancreas (II) was without effect on the CCK concentrations, but decreased the chromogranin A concentration in plasma slightly. The subsequent hemihepatectomy in 2003 that included the metastatic processes (III) normalized plasma CCK and chromogranin A concentrations. The lower panel shows the variations in bodyweight: after an initial decrease the weight increased rapidly during 1999 after onset of chemotherapy. A new period of weight loss accompanied the recurrence of the diarrheas and the gradual increase in plasma CCK and chromogranin A concentrations. Finally, after hemihepatectomy with normalization of CCK in plasma and absence of diarrheas, the weight increased again. Recurrence of liver metastases in 2004-5 resulted again in increased plasma chromogranin A concentrations, but had no effect on CCK production. From ref. Citation17 with permission.
Measurement of CCK has so far not been included in the examination of patients with neuroendocrine tumors because the existence of CCKomas has not been known and because only a few reliable plasma CCK assays are available. Using a highly specific radioimmunoassay for sulfated CCK peptides [Citation17], however, we examined plasma from the patient. Her CCK concentration was 460 pmol/l in the fasting state, which is about 500-fold above normal levels. Such a high concentration of endogenous CCK has never been measured in a patient before. Subsequent measurements of all samples available from the patient showed variations as illustrated in . Gel chromatography showed that the bioactive CCK-forms in plasma corresponded to CCK-83, −58, −33, and −8 (, upper panel). An assay for human proCCK revealed increased concentrations of proCCK and N-terminally truncated fragments of proCCK (, lower panel). It is general knowledge that endocrine tumors often process hormone precursors inefficiently and therefore release increased amounts of the prohormone and immature processing intermediates. Finally, an assay for nonsulfated CCK peptides showed that plasma from the patient contained more nonsulfated than sulfated CCK.
Figure 2. Bioactive CCK and proCCK forms in a plasma sample from the CCKoma patient: by size-chromatography on a calibrated sephadex G50 superfine column, four peptides eluted like CCK-83, -58, -33, and -8 using a specific radioimmunoassay for bioactive, amidated CCK [Citation29]. the assay measures the CCK-forms with equimolar potency without cross-reactivity with homologous gastrin peptides (upper panel). A radioimmunoassay for human proCCK identified in the same plasma sample intact proCCK and three N-terminally truncated forms of proCCK (lower panel). Note the 10-fold higher concentrations of proCCK fragment in the lower panel. From ref. [Citation17] with permission.
![Figure 2. Bioactive CCK and proCCK forms in a plasma sample from the CCKoma patient: by size-chromatography on a calibrated sephadex G50 superfine column, four peptides eluted like CCK-83, -58, -33, and -8 using a specific radioimmunoassay for bioactive, amidated CCK [Citation29]. the assay measures the CCK-forms with equimolar potency without cross-reactivity with homologous gastrin peptides (upper panel). A radioimmunoassay for human proCCK identified in the same plasma sample intact proCCK and three N-terminally truncated forms of proCCK (lower panel). Note the 10-fold higher concentrations of proCCK fragment in the lower panel. From ref. [Citation17] with permission.](/cms/asset/5c053ecc-3069-4a45-8289-09284624ab6c/igas_a_2308532_f0002_b.jpg)
Immunohistochemical examination of the endocrine pancreatic tumor showed one small focus with CCK-containing cells. In contrast, almost all cells in the resected liver metastases contained immunoreactive CCK.
The biology of CCK as a gut hormone
In the normal gastrointestinal tract, CCK is expressed in endocrine I-cells in the small intestinal mucosa [Citation4]. The I-cell density is highest in the duodenal and proximal jejunal mucosa. Since the duodenum is short, however, by far most CCK is synthetized in the jejunum [Citation20,Citation21]. Moreover, in spite of lower I-cell density, even the ileum expresses more CCK than the duodenum. In addition, the mucosa of the colon may sporadically harbor a few I-cells [Citation22,Citation23].
The I-cells mature proCCK to six bioactive CCK-peptides of which five have the same α-amidated and tyrosyl O-sulfated C-terminal heptapeptide sequence (-Tyr(SO4H)-Met-Gly-Trp-Met-Asp-PheNH2) [Citation24]. The short, unsulfated CCK-5 corresponds to the very C-terminal pentapeptide of this sequence (see also and ). The heptapeptide sequence constitutes the epitope for binding to CCK-receptors and is hence the active “site” of the hormone (for extensive review about CCK receptors, see ref. [Citation25]). The CCK-A receptor (the alimentary receptor) is highly specific and requires O-sulfation of the C-terminal Tyr-residue in CCK, whereas the CCK-B receptor (the brain receptor) is more promiscuous and binds both sulfated and nonsulfated CCK peptides (including CCK-5) as well as the homologous gastrin peptides () that circulate in ten-fold higher concentrations than CCK in normal mammals. The CCK-A receptor is expressed fairly abundantly throughout the gastrointestinal mucosa, on gallbladder myocytes, in the pancreas on both exocrine and endocrine cells, and on afferent vagal neurons. In contrast, the CCK-B receptor is widespread in the brain, but additionally expressed on the ECL-cells in the stomach [Citation26] and on islet-cells in the pancreas [Citation27].
Figure 3. Scheme of structurally identified α-amidated products of proCCK. The cleavage sites at basic amino acid residues (R (arginine) and K (lysine)) are indicated on proCCK. s and ns mean ‘sulfated’ and ‘nonsulfated’ respectively.
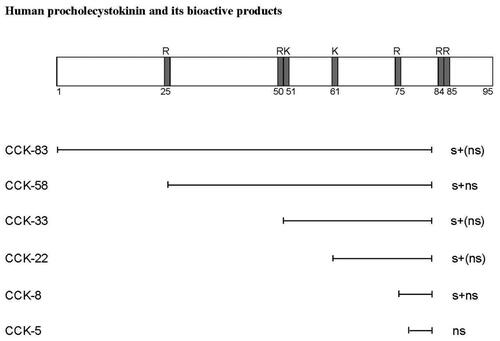
Figure 4. The primary structure of the C-terminal heptapeptide amide sequence that constitutes the specific receptor epitope of bioactive CCK peptides. The corresponding gastrin sequence is aligned below. Note the difference in the position of the tyrosyl residues between CCK and gastrin peptides.
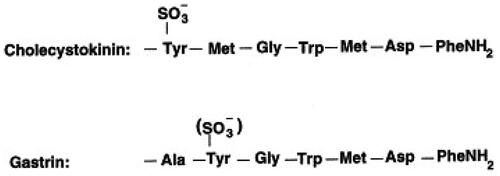
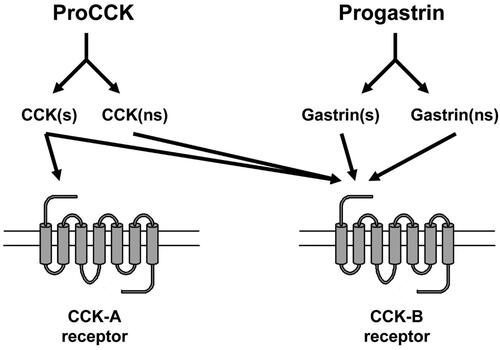
Figure 5. The homologous hormone precursors proCCK and progastrin are both processed to sulfated (s) and nonsulfated (ns) bioactive peptides. Only sulfated CCK peptides are, however, bound with high affinity to the CCK-A receptor, whereas the CCK-B receptor binds all the α-amidated peptide products of both proCCK and progastrin.
Essentially, all CCK peptides in plasma are derived from the enteroendocrine I-cells secreted as a mixture of CCK-58, -33, -22, and -8 [Citation28]. In healthy subjects, the plasma concentrations of bioactive CCK peptides in the fasting state vary individually between 0.1 and 2.0 pmol/l. The concentration peaks 10-15 min after a mixed meal at a level of 4 to 10 pmol/l and then decreases slowly during the next 2-3 h. The secretory pattern depends on the size of the meal and its composition [Citation28–30]. The postprandial increase is sufficient for intestinal O-sulfated CCK to activate a number of digestive functions: Gallbladder contraction; pancreatic enzyme secretion; pancreatic hormone secretion; intestinal enzyme secretion and motility. CCK also contributes to inhibiting gastric secretion and emptying and is the first intestinal satiety signal to be sent to the brain via afferent vagal neurons (for further details, see refs. [4, Citation31–33]).
In terms of gut hormone function, it is, however, decisive to understand that the common textbook concept of “one hormone, one target” is misleading. On the contrary, gut hormones interact in a differentiated way as previously described [Citation34,Citation35]. For instance, CCK and secretin potentiate each other in the stimulation of pancreatic enzyme and bicarbonate secretion [Citation36,Citation37]; in satiety signaling to the hypothalamus, CCK cooperates with GLP-1 (glucagon-like peptide 1) and PYY (peptide tyrosine-tyrosine) in a time-sequential manner [Citation31,Citation38]; and by inhibition of gastric acid secretion and emptying, CCK interacts with secretin, GIP (gastric inhibitory polypeptide), and GLP-1 [Citation39–41].
The processing of proCCK in normal entero-endocrine cells and tumor cells
The normal intestinal I-cells mature as mentioned proCCK to five O-sulfated CCK-peptides (), which all are full agonists for both the CCK-A receptor and the CCK-B receptor [Citation24]. Moreover, also the short, unsulfated C-terminal α-amidated fragment, CCK-5, is produced in low amounts [Citation42]. CCK-5, however, is an agonist only for the CCK-B receptor. The I-cells also contain small amounts of intact proCCK as well as C-terminally truncated processing intermediates that are not bound to any receptor, and hence biologically inactive [Citation43]. Finally, normal I-cells also produce non-sulfated but α-amidated CCK-peptides in amounts corresponding to approximately 25% of the O-sulfated CCKs [Citation20]. These amidated, but unsulfated CCK-peptides are – like CCK-5 – agonists for only the CCK-B receptor [Citation44,Citation45]. Thus, in I-cells, proCCK is processed to multiple fragments [Citation24,Citation46] of which the carboxyamidated peptides are all agonists for the CCK-B receptor (), but only those that, in addition, are tyrosyl O-sulfated become agonists for the CCK-A receptor (i.e., sulfated CCK-83, −58, −33, −22, and −8). This difference in receptor preference () is essential for understanding the pathophysiology of the CCKoma syndrome.
In contrast to normal endocrine cells, endocrine tumor cells often secrete a different molecular pattern with distinctive forms and quantities of bioactive hormones (Citation47–49). These differences correspond to the symptoms manifested by patients with a tumor. shows plasma chromatography of the various CCK peptides found in our CCKoma patient. Accordingly, in the case of a human CCKoma syndrome, the concentration of bioactive CCK forms (i.e., both α-amidated and O-sulfated) in plasma was 500- to 1000-fold higher than normal concentrations [Citation16,Citation17]. Moreover, the chromatographic pattern of the bioactive forms differed, so that also CCK-83 was present in significant amounts (undetectable in normal plasma), whereas CCK-22 was absent (probably due to lack of prohormone convertase 2 in the tumor cells) (see , upper panel). Finally, plasma contained excessive amounts of intact proCCK, and biologically inert processing intermediates [Citation17,Citation43] (see also , lower panel). The specific molecular CCK-pattern in plasma from the CCKoma patient explains to a large degree her symptoms.
The CCKoma symptoms
Rats and mice bearing a transplanted CCK and GLP-1 secreting pancreas tumor developed anorexia, weight loss, and cachexia resulting in death after 5-6 weeks. Their tumor cells produced both bioactive CCK and truncated GLP-1 [Citation14,Citation15]. It is likely that this particular combination of CCK and GLP-1 in high doses is not only a powerful satiety signal [Citation38], but also a lethal starvation mixture. The rodent CCKoma phenotype supports an earlier prediction that CCKomas in man with significant hyperCCKemia would have ‘persistent gallbladder contraction, diarrhea, anorexia, and gastric acid hypersecretion with ensuing duodenal ulcers’ [Citation47]. This prediction has turned out to be correct with the discovery of CCKoma syndrome [Citation16,Citation17]: Hence, the patient displayed four major disorders: Severe non-watery diarrheas, which presumably in combination with low appetite led to a weight loss of almost 20 kg (from 75 to 56 kg) in less than six months. Moreover, she had repeated bile stone attacks from a permanently contracted gallbladder that required cholecystectomy. Finally, she developed recurrent duodenal peptic ulcers (occasionally bleeding) that were treated with proton pump inhibitors. The intensity of the symptoms varied synchronously with the variations in the plasma concentrations of the bioactive CCK peptides [Citation17] (see also ).
Notably, recurrent peptic ulcer disease and diarrhea are symptoms also in the Zollinger-Ellison syndrome known as the phenotype of gastrinomas (gastrin-producing endocrine tumors) [Citation47,Citation50,Citation51]. Gastrin peptides share the CCK-B receptor with CCK peptides (). Gastrin was nevertheless not involved in the CCKoma symptomatology of our patient, because gastrin in plasma – as measured with a reliable gastrin assay [Citation52,Citation53] – remained in normal range concentrations throughout the disease-period of the CCKoma patient [Citation17]. Thus, the actual Zollinger-Ellison symptoms in the CCKoma syndrome are due to the high plasma concentrations of proCCK-derived CCK-B receptor agonists such as CCK-83, −58, −33, −22, and −8 (all in sulfated as well as in non-sulfated molecular forms) including also perhaps CCK-5. Notably, the CCK-B receptor is identical to the originally identified gastrin receptor [Citation54]. The high concentration levels of CCK-peptides therefore stimulated hyper-secretion of gastric acid, which is responsible for the occurrence of duodenal peptic ulcers and contributes to the development of diarrheas.
It looks like a paradox that sulfated CCK peptides as such inhibit gastric acid secretion under physiological circumstances in mammals. The inhibition occurs via the CCK-A receptor expressed on paracrine somatostatin cells neighboring the acid-producing parietal-cells in the gastric fundic mucosa [Citation39,Citation55–57]. The inhibition constitutes the normal enterogastrone mechanism of CCK. Therefore, inhibition of gastric acid secretion might be expected in CCKoma patients. However, the enterogastrone effect of sulfated CCK is apparently overridden by massive amounts of non-sulfated CCK peptides that are agonists only for the CCK-B receptor on gastric ECL-cells, which release histamine that subsequently stimulates the acid secretion from parietal cells [Citation26,Citation58]. The existence of non-sulfated CCK-peptides, including also CCK-5 and their CCK-B receptor agonism, has been widely ignored [Citation44]. However, non-sulfated CCK peptides constitute a separate hormone system in its own right as demonstrated by their specific role in the CCKoma symptoms. The syndrome also illustrates that O-sulfation of peptide hormones is often decreased in tumor cells, as previously demonstrated for gastrin in gastrinomas and in hypersecreting antral G-cells [Citation53,Citation59–61].
The diagnostic challenges of CCKomas
The first premise for correct diagnosis is awareness of the existence of CCKomas – irrespective of whether you are a general practitioner, an endocrine oncologist or a gastroenterologist. Being aware, one should suspect the possibility in tumor patients displaying a combination of the described cardinal symptoms: severe non-watery diarrhea and weight loss; reduced appetite; gallbladder symptoms (bile stones and contraction); and peptic duodenal ulcer disease.
Second, the clinical suspicion of a CCKoma requires confirmation by measurements of CCK and gastrin in plasma from the patient in the fasting state. Due to the homology between CCK and gastrin peptides (), and due to the occurrence of non-sulfated CCK it is necessary that the immunoassays (radioimmunoassay (RIA) or enzyme-linked immunosorbent assay (ELISA)) employed are entirely specific. The CCK assay has to be without any cross-reactivity with gastrin peptides; and likewise, the gastrin assay has to be without any cross-reactivity with CCK peptides – sulfated or unsulfated. In this context, it is a major problem that the close structural homology between CCK and gastrin often renders antibodies raised against gastrin react also with CCK peptides [Citation29,Citation62]. This specificity problem has rarely been examined in relation to unsulfated CCK peptides, a default often observed for commercial gastrinkits [Citation53]. Neglect of the CCK/gastrin specificity problem in diagnostic assays has consequences. Hence, using gastrin assays that cross-react even moderately with CCK makes it likely that a number of CCKoma patients with Zollinger-Ellison symptoms are falsely categorized as gastrinoma patients. Moreover, since 1–3% of Zollinger–Ellison patients have normal gastrin levels [Citation51,Citation53], measurement of plasma concentrations of both sulfated and non-sulfated CCK may provide clarification. Hence, the diagnosis of CCKomas requires assays specific for sulfated CCK peptides, for nonsulfated CCK peptides, and for gastrin. In addition, a processing-independent assay (PIA) for proCCK [Citation48, Citation63–65] may be useful in the quantitation of the entire CCK peptide expression of a CCKoma in the primary tumor and possible metastases, as well as in plasma. If the assay results support a CCKoma suspicion; scannings for localization of the primary tumor and possible metastases are indicated. Notably, treatment with CCK-receptor antagonists has thus far not been examined.
Unrelated to specificity problems of hormone measurements, the latest decades have witnessed another diagnostic problem due to the increasing and widespread intake of proton pump inhibitors (PPIs). Thus, even mild dyspeptic symptoms – as seen also in early phases of the gastrinoma and CCKoma diseases – are treated with PPIs, which masks Zollinger-Ellison symptoms to a degree where only few gastrinoma cases today are discovered [Citation66–71]. Moreover, to add insult to injury, PPIs not only mask clinical symptoms, they also increase antral gastrin secretion to a degree that may overshadow the hypergastrinemia caused by gastrinoma cells. The PPI-induced hypersecretion of gastrin is generally modest, however [Citation72], so that the plasma concentration of gastrin often remains within the normal reference range as seen also in our CCKoma patient [Citation17]. Furthermore, our CCKoma patient was PPI-treated in two periods only.
Measurement of chromogranin A is a useful marker for neuroendocrine tumors in two respects: It supports the neuroendocrine diagnosis of a tumor, and it is most often efficient in monitoring neuroendocrine tumor growth and the effects of therapeutic interventions [Citation73–76] as demonstrated also in our CCKoma patient ( and ref. [Citation17]). Chromogranin A measurements, however, do not differentiate between the specific CCKomas, gastrinomas, and other neuroendocrine tumors.
Further on accurate measurement of CCK and gastrin for CCKoma diagnosis
Suspicion of a CCKoma syndrome requires, as mentioned, confirmation by measurement of CCK and gastrin concentrations in plasma. Since only few CCK assays are available for reliable quantitation of plasma concentrations [Citation29], an expedient first step is accurate measurement of gastrin in plasma or serum [Citation77]. Gastrin should be measured if a CCKoma is suspected, because far most Zollinger-Ellison syndromes are caused by hypersecretion from gastrinomas [Citation18,Citation19,Citation51], because gastrin peptides are homologues to CCK () and because gastrin shares agonism for the CCK-B receptor with CCK peptides (). Like CCK, however, gastrin also circulates in different bioactive forms: Gastrin-71, Gastrin − 34, Gastrin − 17, Gastrin − 14, and Gastrin − 6; the four larger forms in both tyrosyl-O-sulfated and non-sulfated forms [Citation77–81]. Hence, the gastrin assay used for diagnostics has to measure the sum of the described sulfated and nonsulfated gastrins. A minimal demand for specificity examination of the gastrin assay is consequently demonstration that the assay measures at least gastrin-34 and gastrin-17 in both sulfated and non-sulfated forms with equimolar potency and without cross-reactivity with CCK (for which examination of sulfated and non-sulfated CCK-8 may suffice). If such gastrin assay measures normal concentration in plasma from a fasting patient with CCKoma symptoms, the necessity for specific CCK quantitation increases and renders the CCKoma diagnosis considerably more likely. Final diagnostic confirmation then requires that a plasma sample is shipped to a CCK-laboratory having sufficient analytical competence and capacity for accurate quantitation of both sulfated and non-sulfated CCK [Citation29]. Unfortunately, most commercial CCK-kits available today are unreliable and have so far not been shown to possess the necessary specificity [Citation82].
Like other peptide-producing tumors, CCKomas release – in addition to the amidated bioactive peptides – also substantial amounts of inactive precursors (in casu proCCK) and processing intermediates [Citation43] to plasma. Therefore, measurement by the processing-independent analysis (PIA) may contribute to characterize the total tumor expression of proCCK-derived fragments (see and refs. [Citation48,Citation65]).
Conclusion
Among neuroendocrine tumor syndromes, the CCKoma syndrome is novel [Citation3]. It is simply a new disease. However, it is striking that only one clear-cut case in humans has been documented so far [Citation16,Citation17]. Equally surprising, this singular case has not been followed up by further identifications of additional CCKoma patients in the last decade since the first description [Citation16]. For the well-established enteropancreatic endocrine tumor syndromes (insulinomas, gastrinomas, glucagonomas, somatostatinomas, and VIPomas), the first descriptions were fairly rapidly followed by reports of additional patient cases [Citation3].
The first casuistic description of a new syndrome or disease may per definition suggest that the syndrome is rare. It is, however, hard to believe that the singular Danish woman in whom we discovered a malignant CCKoma [Citation16,Citation17], would be entirely unique. More likely, some CCKoma patients are camouflaged under a wrong diagnosis. For two reasons it would be obvious to assume that some CCKoma patients are miscategorized as gastrin’ergic Zollinger-Ellison patients. Thus, the combination of similar clinical symptoms and the fact that several gastrin-assays on the market are short of the necessary specificity might easily explain the misdiagnosis.
Proper diagnosis requires CCK and gastrin assays whose specificity is carefully examined. It is difficult to develop antibodies of sufficient affinity that also are entirely specific for CCK peptides. Nevertheless, it can be done [Citation29,Citation83]. Moreover, it is likely that laboratories running specific CCK and gastrin assays will be open for assistance with the necessary diagnostic measurements. A final wish in a CCKoma context is that Neuroendocrine Tumor Societies would describe – or at least mention – the existence of CCKomas in their guidance papers [Citation84].
Acknowledgement
The highly skilled and patient secretarial assistance of Connie Bundgaard (MA) is most gratefully acknowledged.
No potential conflict of interest was reported by the author.
Disclosure statement
Additional information
Funding
References
- Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9(1):61–72. doi: 10.1016/S1470-2045(07)70410-2.
- Lawrence B, Gustafsson BI, Chan A, et al. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40(1):1–18, vii. doi: 10.1016/j.ecl.2010.12.005.
- de Herder WW, Rehfeld JF, Kidd M, et al. A short history of neuroendocrine tumours and their peptide hormones. Best Pract Res Clin Endocrinol Metab. 2016;30(1):3–17. doi: 10.1016/j.beem.2015.10.004.
- Rehfeld JF. Cholecystokinin and the hormone concept. Endocr Connect. 2021;10(3):R139–R150. doi: 10.1530/EC-21-0025.
- Rehfeld JF. Immunochemical studies on cholecystokinin. II. Distribution and molecular heterogeneity in the Central nervous system and small intestine of man and hog. J Biol Chem. 1978;253(11):4022–4030. doi: 10.1016/S0021-9258(17)34793-2.
- Larsson LI, Rehfeld JF. Localization and molecular heterogeneity of cholecystokinin in the Central and peripheral nervous system. Brain Res. 1979;165(2):201–218. doi: 10.1016/0006-8993(79)90554-7.
- Rehfeld JF, Hansen HF. Characterization of preprocholecystokinin products in the porcine cerebral cortex. Evidence of different processing pathways. J Biol Chem. 1986;261(13):5832–5840.
- Rehfeld JF, Lindholm J, Andersen BN, et al. Pituitary tumors containing cholecystokinin. N Engl J Med. 1987;316(20):1244–1247. doi: 10.1056/NEJM198705143162004.
- Rehfeld JF, Johnsen AH, Odum L, et al. Non-sulfated cholecystokinin in human medullary thyroid carcinomas. J Endocrinol. 1990;124(3):501–506. doi: 10.1677/joe.0.1240501.
- Bardram L, Hilsted L, Rehfeld JF. Cholecystokinin, gastrin and their precursors in pheochromocytomas. Acta Endocrinol (Copenh). 1989;120(4):479–484. doi: 10.1530/acta.0.1200479.
- Reubi JC, Koefoed P, Hansen T, et al. Procholecystokinin as marker of human Ewing sarcomas. Clin Cancer Res. 2004;10(16):5523–5530. doi: 10.1158/1078-0432.CCR-1015-03.
- Oikonomou E, Buchfelder M, Adams EF. Cholecystokinin (CCK) and CCK receptor expression by human gliomas: evidence for an autocrine/paracrine stimulatory loop. Neuropeptides. 2008;42(3):255–265. doi: 10.1016/j.npep.2008.02.005.
- Rehfeld JF, van Solinge WW, Tos M, et al. Gastrin, cholecystokinin and their precursors in acoustic neuromas. Brain Res. 1990;530(2):235–238. doi: 10.1016/0006-8993(90)91288-r.
- Madsen OD, Larsson LI, Rehfeld JF, et al. Cloned cell lines from a transplantable islet cell tumor are heterogeneous and express cholecystokinin in addition to islet hormones. J Cell Biol. 1986;103(5):2025–2034. doi: 10.1083/jcb.103.5.2025.
- Madsen OD, Karlsen C, Nielsen E, et al. The dissociation of tumor-induced weight loss from hypoglycemia in a transplantable pluripotent rat islet tumor results in the segregation of stable alpha- and beta-cell tumor phenotypes. Endocrinology. 1993;133(5):2022–2030. doi: 10.1210/endo.133.5.8404649.
- Rehfeld JF, Federspiel B, Bardram L. A neuroendocrine tumor syndrome from cholecystokinin secretion. N Engl J Med. 2013;368(12):1165–1166. doi: 10.1056/NEJMc1215137.
- Rehfeld JF, Federspiel B, Agersnap M, et al. The uncovering and characterization of a CCKoma syndrome in enteropancreatic neuroendocrine tumor patients. Scand J Gastroenterol. 2016;51(10):1172–1178. doi: 10.1080/00365521.2016.1183706.
- Jensen RT. Gastrinoma as a model for prolonged hypergastrinemia in man. In Walsh JH, editor. Gastrin. New York: Raven Press Publishing Co. 1993:pp. 373–393.
- Jensen RT. Recent insights from studies of gastrinomas. In: merchant JL, Buchan AMJ, Wang TC, editors. Gastrin in the new millenium. CURE Foundation: Los Angeles, CA, USA, 2004:pp. 339–352.
- Agersnap M, Rehfeld JF. Nonsulfated cholecystokinins in the small intestine of pigs and rats. Peptides. 2015;71:121–127. doi: 10.1016/j.peptides.2015.07.010.
- Gilliam-Vigh H, Jorsal T, Rehfeld JF, et al. Expression of cholecystokinin and its receptors in the intestinal tract of type 2 diabetes patients and healthy controls. J Clin Endocrinol Metab. 2021;106(8):2164–2170. doi: 10.1210/clinem/dgab367.
- Martins P, Fakhry J, de Oliveira EC, et al. Analysis of enteroendocrine cell populations in the human Colon. Cell Tissue Res. 2017;367(2):161–168. doi: 10.1007/s00441-016-2530-7.
- Fakhry J, Wang J, Martins P, et al. Distribution and characterisation of CCK containing enteroendocrine cells of the mouse small and large intestine. Cell Tissue Res. 2017;369(2):245–253. doi: 10.1007/s00441-017-2612-1.
- Rehfeld JF. The endoproteolytic maturation of progastrin and procholecystokinin. J Mol Med (Berl). 2006;84(7):544–550. doi: 10.1007/s00109-006-0055-3.
- Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86(3):805–847. doi: 10.1152/physrev.00014.2005.
- Ding XQ, Lindström E, Håkanson R. Evaluation of three novel cholecystokinin-B/gastrin receptor antagonists: a study of their effects on rat stomach enterochromaffin-like cell activity. Pharmacol Toxicol. 1997;81(5):232–237. doi: 10.1111/j.1600-0773.1997.tb00052.x.
- Reubi JC, Waser B, Gugger M, et al. Distribution of CCK1 and CCK2 receptors in normal and diseased human pancreatic tissue. Gastroenterology. 2003;125(1):98–106. doi: 10.1016/s0016-5085(03)00697-8.
- Rehfeld JF, Sun G, Christensen T, et al. The predominant cholecystokinin in human plasma and intestine is cholecystokinin-33. J Clin Endocrinol Metab. 2001;86(1):251–258. doi: 10.1210/jc.86.1.251.
- Rehfeld JF. Accurate measurement of cholecystokinin in plasma. Clin Chem. 1998;44(5):991–1001. doi: 10.1093/clinchem/44.5.991.
- Veedfald S, Rehfeld JF, van Hall G, et al. Entero-pancreatic hormone secretion, gastric emptying, and glucose absorption after frequently sampled meal tests. J Clin Endocrinol Metab. 2022;107(1):e188–e204. doi: 10.1210/clinem/dgab610.
- Rehfeld JF. Cholecystokinin. In Maklouf GM, handbook of physiology, section 6., vol. II. Amer Physiol Soc., Bethesda (Md), 1989:pp. 337–358.
- Liddle RA. Cholecystokinin. In: Walsh JH, Dockray GJ, editors. Gut peptides: biochemistry and physiology. Raven Press Ltd., New York, 1994:pp. 175–216.
- Rehfeld JF. Cholecystokinin – from local gut hormone to ubiquitous messenger. Front Endocrinol (Lausanne). 2017;8art. :47. doi: 10.3389/fendo.2017.00047.
- Rehfeld JF. Incretin physiology beyond glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide: cholecystokinin and gastrin peptides. Acta Physiol (Oxf). 2011;201(4):405–411. doi: 10.1111/j.1748-1716.2010.02235.x.
- Rehfeld JF. The origin and understanding of the incretin concept. Front Endocrinol (Lausanne). 2018;9art. :387. doi: 10.3389/fendo.2018.00387.
- Henriksen FW, Worning H. The interaction of secretin and pancreozymin on the exocrine pancreatic secretion in dogs. Acta Physiol Scand. 1969;76(1):67–72. doi: 10.1111/j.1748-1716.1969.tb04451.x.
- Wormsley KGA. A comparison of the response to secretin, pancreozymin and a combination of these hormones in man. Scand J Gastroenterol. 1969;4(5):413–417. doi: 10.3109/00365526909180626.
- Vana V, Laerke MK, Rehfeld JF, et al. Vagal afferent cholecystokinin receptor activation is required for glucagon-like peptide-1-induced satiation. Diabetes Obes Metab. 2022;24(2):268–280. doi: 10.1111/dom.14575.
- Chen D, Zhao CM, Håkanson R, et al. Altered control of gastric acid secretion in gastrin-cholecystokinin double mutant mice. Gastroenterology. 2004;126(2):476–487. doi: 10.1053/j.gastro.2003.11.012.
- Asmar M, Bache M, Knop FK, et al. Do the actions of glucagon-like peptide-1 on gastric emptying, appetite, and food intake involve release of amylin in humans? J Clin Endocrinol Metab. 2010;95(5):2367–2375. doi: 10.1210/jc.2009-2133.
- Brown JC. A gastric inhibitory polypeptide. I. The amino acid composition and the tryptic peptides. Can J Biochem. 1971;49(2):255–261. doi: 10.1139/o71-037.
- Shively J, Reeve JR, Jr, Eysselein VE, et al. CCK-5: sequence analysis of a small cholecystokinin from canine brain and intestine. Am J Physiol. 1987;252(2 Pt 1):G272–G275. doi: 10.1152/ajpgi.1987.252.2.G272.
- Cantor P, Rehfeld JF. Cholecystokinin in pig plasma: release of components devoid of a bioactive COOH-terminus. Am J Physiol. 1989;256(1 Pt 1):G53–G61. doi: 10.1152/ajpgi.1989.256.1.G53.
- Rehfeld JF, Agersnap M. Unsulfated cholecystokinin: an overlooked hormone? Regul Pept. 2012;173(1-3):1–5. doi: 10.1016/j.regpep.2011.09.009.
- Agersnap M, Rehfeld JF. Measurement of nonsulfated cholecystokinins. Scand J Clin Lab Invest. 2014;74(5):424–431. doi: 10.3109/00365513.2014.900695.
- Rehfeld JF, Bundgaard JR, Hannibal J, et al. The cell-specific pattern of cholecystokinin peptides in endocrine cells versus neurons is governed by the expression of prohormone convertases 1/3, 2, and 5/6. Endocrinology. 2008;149(4):1600–1608. doi: 10.1210/en.2007-0278.
- Rehfeld JF, van Solinge WW. The tumor biology of gastrin and cholecystokinin. Adv Cancer Res. 1994;63:295–347. doi: 10.1016/s0065-230x(08)60403-0.
- Paloheimo LI, Rehfeld JF. Quantitation of procholecystokinin and its products in plasma by processing-independent analysis. Clin Chim Acta. 1995;238(1):21–33. doi: 10.1016/0009-8981(95)06055-i.
- Rehfeld JF. Cholecystokinin expression in tumors: biogenetic and diagnostic implications. Future Oncol. 2016;12(18):2135–2147. doi: 10.2217/fon-2015-0053.
- Zollinger RM, Ellison EH. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann Surg. 1955;142(4):709–728.
- Wolfe MM, Jensen RT. Zollinger-Ellison syndrome. Current concepts in diagnosis and management. N Engl J Med. 1987;317(19):1200–1209. doi: 10.1056/NEJM198711053171907.
- Stadil F, Rehfeld JF. Determination of gastrin in serum. Eval Reliability Radioimmunoassay. Scand J Gastroenterol. 1973;8(2):101–112. doi: 10.1080/00365521.1973.12096677.
- Rehfeld JF, Gingras MH, Bardram L, et al. The Zollinger-Ellison syndrome and mismeasurement of gastrin. Gastroenterology. 2011;140(5):1444–1453. doi: 10.1053/j.gastro.2011.01.051.
- Kopin AS, Lee YM, McBride EW, et al. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc Natl Acad Sci U S A. 1992;89(8):3605–3609. doi: 10.1073/pnas.89.8.3605.
- Larsson LI, Goltermann N, de Magistris L, et al. Somatostatin cell processes as pathways for paracrine secretion. Science. 1979;205(4413):1393–1395. doi: 10.1126/science.382360.
- Makhlouf GM, Schubert ML. Gastric somatostatin: a paracrine regulator of acid secretion. Metabolism. 1990;39(9 Suppl 2):138–142. doi: 10.1016/0026-0495(90)90232-2.
- Lloyd KC, Raybould HE, Walsh JH. Cholecystokinin inhibits gastric acid secretion through type "a" cholecystokinin receptors and somatostatin in rats. Am J Physiol. 1992;263(3 Pt 1):G287–G292. doi: 10.1152/ajpgi.1992.263.3.G287.
- Chen D, Zhao CM, Al-Haider W, et al. Differentiation of gastric ECL cells is altered in CCK(2) receptor-deficient mice. Gastroenterology. 2002;123(2):577–585. doi: 10.1053/gast.2002.34746.
- Andersen BN, Stadil F. Sulfation of gastrin in zollinger–ellison sera: evidence for association between sulfation and proteolytic processing. Regul Pept. 1983;6(3):231–239. doi: 10.1016/0167-0115(83)90141-6.
- Andersen BN, Petersen B, Borch K, et al. Variations in the sulfation of circulating gastrins in gastrointestinal diseases. Scand J Gastroenterol. 1983;18(4):565–569. doi: 10.3109/00365528309181639.
- Jensen S, Borch K, Hilsted L, et al. Progastrin processing during antral G-cell hypersecretion in humans. Gastroenterology. 1989;96(4):1063–1070. doi: 10.1016/0016-5085(89)91624-7.
- Rehfeld JF, Stadil F, Rubin B. Production and evaluation of antibodies for the radioimmunoassay of gastrin. Scand J Clin Lab Invest. 1972;30(2):221–232. doi: 10.3109/00365517209081114.
- Bardram L, Rehfeld JF. Processing-independent radioimmunoanalysis: a general analytical principle applied to progastrin and its products. Anal Biochem. 1988;175(2):537–543. doi: 10.1016/0003-2697(88)90580-5.
- Rehfeld JF, Goetze JP. The posttranslational phase of gene expression: new possibilities in molecular diagnosis. Curr Mol Med. 2003;3(1):25–38. doi: 10.2174/1566524033361717.
- Rehfeld JF, Goetze JP. Processing-independent analysis (PIA): a method for quantitation of the total peptide-gene expression. Peptides. 2021;135:170427. doi: 10.1016/j.peptides.2020.170427.
- Corleto VD, Annibale B, Gibril F, et al. Does the widespread use of proton pump inhibitors mask, complicate and/or delay the diagnosis of Zollinger-Ellison syndrome? Aliment Pharmacol Ther. 2001;15(10):1555–1561. doi: 10.1046/j.1365-2036.2001.01085.x.
- Ito T, Cadiot G, Jensen RT. Diagnosis of zollinger ellison syndrome: increasingly difficult. World J Gastroenterol. 2012;18(39):5495–5503. doi: 10.3748/wjg.v18.i39.5495.
- Metz DC. Diagnosis of the Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2012;10(2):126–130. doi: 10.1016/j.cgh.2011.07.012.
- Raines D, Chester M, Diebold AE, et al. A prospective evaluation of the effect of chronic proton pump inhibitor use on plasma biomarker levels in humans. Pancreas. 2012;41(4):508–511. doi: 10.1097/MPA.0b013e318243a0b6.
- Metz DC, Cadiot G, Poitras P, et al. Diagnosis of Zollinger-Ellison syndrome in the era of PPIs, faulty gastrin assays, sensitive imaging and limited access to acid secretory testing. Int J Endocr Oncol. 2017;4(4):167–185. doi: 10.2217/ije-2017-0018.
- Rossi RE, Elvevi A, Citterio D, et al. Gastrinoma and Zollinger-Ellison syndrome: a roadmap for the management between new and old therapies. World J Gastroenterol. 2021;27(35):5890–5907. doi: 10.3748/wjg.v27.i35.5890.
- Rehfeld JF. Gastrin and the moderate hypergastrinemias. Int J Mol Sci. 2021;22(13):6977. doi: 10.3390/ijms22136977.
- Børglum T, Rehfeld JF, Drivsholm LB, et al. Processing-independent quantitation of chromogranin A in plasma from patients with neuroendocriine tumors and small-cell lung carcinomas. Clin Chem. 2007;53(3):438–446. doi: 10.1373/clinchem.2006.076158.
- Massironi S, Rossi RE, Casazza G, et al. Chromogranin A in diagnosing and monitoring patients with gastroenteropancreatic neuroendocrine neoplasms: a large series from a single institution. Neuroendocrinology. 2014;100(2-3):240–249. doi: 10.1159/000369818.
- Rossi RE, Ciafardini C, Sciola V, et al. Chromogranin A in the follow-up of gastroenteropancratic neuroendocrine neoplasms. Is it really game over? A systematic review and meta-analysis. Pancreas. 2018;47(10):1249–1255. doi: 10.1097/MPA.0000000000001184.
- Rehfeld JF, Broedbaek K, Goetze JP, et al. True Chromogranin A concentrations from patients with small intestinal neuroendocrine tumours. Scand J Gastroenterol. 2020;55(5):565–573. doi: 10.1080/00365521.2020.1759141.
- Rehfeld JF. The art of measuring gastrin in plasma: a dwindling diagnostic discipline? Scand J Clin Lab Invest. 2008;68(5):353–361. doi: 10.1080/00365510701771831.
- Rehfeld JF, Stadil F, Vikelsoe J. Immunoreactive gastrin components in human serum. Gut. 1974;15(2):102–111. doi: 10.1136/gut.15.2.102.
- Rehfeld JF, Johnsen AH. Identification of gastrin component I as gastrin-71. The largest possible bioactive progastrin product. Eur J Biochem. 1994;223(3):765–773. doi: 10.1111/j.1432-1033.1994.tb19051.x.
- Rehfeld JF, Hansen CP, Johnsen AH. Post-poly(glu) cleavage and degradation modified by O-sulfated tyrosine: a novel post-translational processing mechanism. Embo J. 1995;14(2):389–396. doi: 10.1002/j.1460-2075.1995.tb07013.x.
- Schubert ML, Rehfeld JF. Gastric peptides-gastrin and somatostatin. Compr Physiol. 2020;10(1):197–228. doi: 10.1002/cphy.c180035.
- Rehfeld JF. Measurement of cholecystokinin in plasma with reference to nutrition related obesity studies. Nutr Res. 2020;76:1–8. doi: 10.1016/j.nutres.2020.01.003.
- Rehfeld JF. How to measure cholecystokinin in tissue, plasma and cerebrospinal fluid. Regul Pept. 1998;78(1-3):31–39. doi: 10.1016/s0167-0115(98)00133-5.
- Hofland J, Falconi M, Christ E, et al. European neuroendocrine tumor society 2023 guidance for functioning pancreatic neuroendocrine tumour syndromes. J Neuroendocrinol. 2023;35(8):e13318. doi: 10.1111/jne.13318.