
Abstract
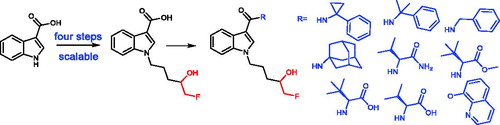
Synthetic cannabinoid metabolites with a 5F-4OH pentyl side chain are of great interest, and several synthetic methods have been developed for the synthesis of these types of metabolites in the literature, which is either lengthy or inappropriate for upscaling. Herein, a straightforward method has been developed and reported for the synthesis of a key intermediate in four steps with an overall yield of 49% on a multigram scale, and its application in the synthesis of nine potential metabolites of synthetic cannabinoids with a 5F-4OH pentyl side chain.
Graphical Abstract
Introduction
Synthetic cannabinoids (SCs) are one of the major groups of new psychoactive substances (NPS) that have been reported to the United Nations Office on Drugs and Crime (UNODC) since 2009.[Citation1] Many of them are used recreationally as replacements to cannabis in order to circumvent legislation. As of January 2020, more than 280 synthetic cannabinoids have been reported to the UNODC Early Warning Advisory (UNODC EWA) from 120 countries and territories.[Citation1] In 2019, synthetic cannabinoids were detected in 28% of the NPS fatality cases that were reported to the UNODC EWA. The potential risk of SCs is much higher. In Japan, 858 SCs were scheduled as narcotics or designated substances as of April 2015.[Citation2] Furthermore, an outbreak of adverse events associated with SCs caused 17 deaths and resulted in 1243 emergency room visits in the State of Mississippi, USA, in April and May 2015.[Citation3] Synthetic cannabinoids are normally metabolized rapidly, which makes their detection challenging especially when the parent substances are not detected.[Citation4-6] The availability of reference standards of the formed metabolites is necessary for the development of analytical methods that lead to the identification of the intake of the parent synthetic cannabinoid. Moreover, the access to metabolite standards is of great interest for metabolism and toxicological studies of SCs.[Citation7-13]
Synthetic cannabinoids have some common structural features, which can be found in .[Citation14,Citation15] Many of them contain a flexible alkyl chain, commonly, a pentyl or 5-fluoro pentyl side chain. It was found that the alkyl chains are metabolized extensively to give mono or di- hydroxylated synthetic cannabinoids as metabolites, and the mono 5-hydroxyl metabolite can be further oxidized to carboxylic acid.[Citation8] Synthetic cannabinoids with a pentyl side chain and their 5-F analogues share quite a few metabolites, which makes it difficult to distinguish between them when the parent drugs are not detectable.[Citation16] It was found by Gamage et al. that 5F-4OH metabolites of five synthetic cannabinoids with an indazole core retain efficacy at the hCB1 receptor, which may contribute to intoxication of abused SCs.[Citation13] Therefore, metabolites with the fluoro group intact are of great interest. Moreover, several synthetic cannabinoids with a 5-F pentyl side chains are among the most frequently reported and featured SCs in fatalities as well as clinical admissions in 2019 according to the UNODC EWA.[Citation1]
Figure 1. General structural information of synthetic cannabinoids with JWH-018 as an example, where the dotted lines are connected bonds. It was adopted from European Monitoring Center for Drugs and Drug Addiction (EMCDDA) and modified.[Citation14,Citation15]
![Figure 1. General structural information of synthetic cannabinoids with JWH-018 as an example, where the dotted lines are connected bonds. It was adopted from European Monitoring Center for Drugs and Drug Addiction (EMCDDA) and modified.[Citation14,Citation15]](/cms/asset/f7283502-e3c9-460b-a10c-0d2471a686ae/lsyc_a_1854786_f0001_c.jpg)
There are few reports on the synthesis of 5F-4OH synthetic cannabinoid metabolites, which vary from three to ten steps (Scheme 1). Moran et al. reported a synthetic route to synthesize the chiral 4OH-AM2201 metabolites with six steps in 11% overall yields.[Citation17] Later in 2018, McKinnie et al. developed a synthetic route to achieve the 4OH metabolite of 5F-APINACA and CUMYL-5F-PINACA in better yields but with ten steps.[Citation18] Considering the rapid change in the illicit drug market, a concise synthetic route has recently been reported for the synthesis of the 5F-4OH metabolites in just three steps.[Citation19] The synthetic route has been shorten, but the disadvantage is that it needs three steps for every single metabolite, and some of the metabolites need to be purified using preparative LC to get the metabolite with high purity (>98%).[Citation19] The method is then not likely used in industrial production. And the access to reference standard from a qualified company, not a normal organic lab is important for forensic society. Considering all these reasons and the rapid change of the illicit market, where a simple replacement of R1 group () may generate a new synthetic cannabinoid, metabolite intermediates which can be used for rapid synthesis of the potential metabolites of synthetic cannabinoids are important. Therefore, a synthetic route for the synthesis of a key intermediate, which subsequently can be used to synthesize the 4OH metabolites of synthetic cannabinoids with an indole core and a 5-F pentyl side chain in only one step was developed. The synthesis method to the intermediate is carried out on a multigram scale with just one of the steps to be purified using silica gel chromatography.
Scheme 1. Overview of synthesis of 5F-4OH synthetic cannabinoid metabolites.

Results and discussion
The 3-indole carboxylic acid 2 (15 grams) was esterified with benzyl bromide under basic condition using Cs2CO3 at room temperature (rt) and yielded 93% (Scheme 2). After the work-up and filtration, the solid was used in next step without further purification. The alkylation of compound 3 with 2-(3-bromopropyl)oxirane using NaH as the base gave a high yield of 83%. The reaction was started in an ice bath and stirred further at rt for about 3 h. The reaction gave the hydrolysis side product of compound 4 with the loss of the benzyl group, which can be removed under basic conditions during work-up. It is worth noting that the attempt of opening the epoxide ring of this side product to achieve compound 1 failed to give a satisfying result using the conditions depicted below. Compound 4 that was achieved from this step, did not require purification by silica gel chromatography either. Two methods were used for the ring opening of epoxide 4. In method A, the epoxide was opened with TBAF in THF under reflux at 80 °C. Product 5 was achieved in a moderate yield of 66%. Another condition (Method B) was also tested using Bu4NH2F3 as the fluorinating reagent in a neat reaction at 85 °C, which was synthesized according to the description by McKinnie et al.[Citation18] The reaction was first attempted at a lower temperature of 60 °C to see whether the formation of side products could be suppressed or not, but it was unsuccessful. The temperature was then increased to 85 °C. This Method B worked as good as Method A, but the reaction time was longer at a similar temperature. The yield was unfortunately not improved. The isomer with ring opening from 4-position was detected under both conditions, where Method B gave slightly more isomer compared to Method A. The advantage of Method B is that the reaction was performed under neat conditions, therefore, no solvents were needed for the reaction. The hydrogenation of 5 with H2 gas and palladium on activated charcoal to remove the protective group went smoothly to produce the key intermediate 1 in a very high yield of 96%.
Scheme 2. Synthesis of the key intermediate 1.

The key intermediate 1 was applied in the synthesis of nine 5F-4OH synthetic cannabinoid metabolites, where they were selected based on their potencies [Citation15,Citation20-22] or availability of reagents (), among which, 5F-CUMYL-PICA and 5F-MDMB-PICA are two of the identified SCs in the intoxication/fatality cases that were reported to the UNDOC EWA in 2019.[Citation1] A common amide coupling condition was used, with TBTU as the additive and TEA as the base. The amide coupling went smoothly to give compounds 6b–f in good yields (Scheme 3). Compound 6a was achieved in a moderate yield because the reaction was stopped before it was completed. The intermediate formed from TBTU and compound 1 had the same retention time as that of the product 6a under the applied analytical liquid chromatography conditions, which caused misinterpretation of the extent of the conversion. However, this intermediate is stable and could be used further to react with amantadine to give the final product 6a. The amid coupling of 1 with benzylamine affording 6c gave good yield after 2 h at rt, which might be due to the less hindrance of a primary amine. The yields of 6g and 6h were moderate due to the formation of bis-coupling side products, where 6g–h coupled further with their respective amino acid. The yields of 6g–h could potentially be improved if they were prepared from their methyl esters, which could be synthesized using this method. In general, the amide coupling step gave high yields and purities of products except for 6g and 6h. Interestingly, although 6e–h are diastereomers, they were not able to be separated under our analytical conditions (Appendix A of the Supplemental part). Probably, it is due to the generated chiral center at the 4-position of the 5F-pentyl side chain is too far from the other chiral center and our conditions were not optimal for their separations. Unlike the amide coupling, the conversion to 6i, an ester, was very slow at 65 °C. Other conditions, such as irradiation under microwave for 45 min at 120 °C and 145 °C were tried. Unfortunately, the conversion of 6i was less than 50%, and the product became less stable. Although the reactions to 6a and 6e–f took longer time at rt, they could be sped up greatly by heating at 65 °C according to the result of 6b and 6d.
Scheme 3. Synthesis of nine potential 5F-4OH synthetic cannabinoid metabolites from key intermediate 1. General conditions: TBTU, TEA, amine/amino acid/alcohol, rt for 2 h to two nights or 65 °C for 2 h to five nights.

Figure 2. Chemical structures of the studied synthetic cannabinoids.

Experimental
General information
HPLC-MS was performed on a Waters system (Column: XSELECT Phenyl-Hexyl, 5 µm, 250 × 19 mm and Waters X-Bridge C-18, 3.5 μm, 50 × 4.6 mm) for preparative and analytical experiments respectively; Mobile phase: water phase A: acetonitrile:water 5:95, with 10 mM NH4OAc; organic phase B: acetonitrile:water 90:10, with 10 mM NH4OAc. Flash chromatography was performed using the following silica gel: High purity grade (Merck Grade 9385), pore size 60 Å, 230–240 mesh particle size. 1H, 19F or 13C-NMR spectra were recorded on a Varian Mercury 300/282/75 MHz or 500/126 MHz instrument (25 °C in CDCl3 or CD3OD). The high-resolution mass measurements were analyzed using Bruker Daltonics UltrafleXtreme MALDI system.
Synthesis of benzyl 1-(5-fluoro-4-hydroxypentyl)-1H-indole-3-carboxylate (5)
Method A:[Citation19] TBAF in THF (27.4 mL, 27.4 mmol) was added to a round bottom flask containing compound 4 (2.301 g, 6.86 mmol), and the mixture was refluxed at 80 °C overnight. The reaction mixture was concentrated, diluted with EtOAc (100 mL), and washed with water (2 × 40 mL). The organic layers were combined and washed with brine (40 mL) and evaporated under reduced pressure to give a crude product (2.9 g). The crude product was purified with silica gel column chromatography using EtOAc:n-heptane (1:2–1:1) to give product 5 (1.62 g, 66%) as a light-yellow sticky oil. 1H-NMR (300 MHz, CDCl3): δ 8.23–8.16 (m, 1H), 7.86 (s, 1H), 7.52–7.25 (m, 8H), 5.40 (s, 2H), 4.49–4.06 (m, 4H), 3.96–3.77 (m, 1H), 2.17–1.93 (m, 2H), 1.52–1.41 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 165.0, 137.0, 136.6, 136.4, 134.5, 128.7, 128.3, 128.1, 126.9, 123.0, 122.1, 122.04, 110.1, 86.7 (d, JCF = 169.2 Hz), 70.1 (d, JCF = 18.8 Hz), 65.7, 46.9, 28.9 (d, JCF = 6.8 Hz), 26.1. 19F NMR (282 MHz, CDCl3): δ − 228.13 (td, JHF = 47.2, 17.3 Hz). HRMS-MALDI (M + Na)+: Calculated for [C21H22FN2O3+Na]+: 378.1481; Found: 378.1475.
Method B:[Citation18] Compound 4 (7.23 g, 21.6 mmol) was combined with tetra-N-butylammonium dihydrogen trifluoride (13.01 g, 43.2 mmol) and the reaction mixture was stirred at 85 °C over two nights. The reaction mixture was dissolved in EtOAc (150 mL) and washed with water (2 × 70 mL). The water layers were combined and extracted with EtOAc (50 mL). The organic layers were combined and washed with sat. NaCl (50 mL). The organic phase was concentrated under vacuum. The crude product was purified as was described in Method A to give compound 5 (4.67 g, 61%) as a light-yellow sticky oil.
Synthesis of 1-(5-fluoro-4-hydroxypentyl)-1H-indole-3-carboxylic acid (1)
Compound 5 (4.70 g, 13.2 mmol) was dissolved in MeOH (150 mL). 10% Pd/C (561 mg, 0.528 mmol) was added, and the reaction mixture was stirred under hydrogen atmosphere overnight (about 22 h). Gravity filtration of the solution through cotton was performed twice. The solution was washed with EtOAc. Concentration of the solution resulted in a pale pink solid (3.35 g, 96%). 1H-NMR (300 MHz, CDCl3) δ 8.27–8.19 (m, 1H), 7.93 (s, 1H), 7.41–7.35 (m, 1H), 7.34–7.27 (m, 2H), 4.48–4.08 (m, 4H), 3.97–3.78 (m, 1H), 2.21–1.93 (m, 2H), 1.54–1.37 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 170.4, 136.8, 135.6, 127.1, 123.1, 122.4, 122.1, 110.2, 106.6, 86.8 (d, JCF = 169.1 Hz), 70.1 (d, JCF = 19.2 Hz), 47.0, 28.9 (d, JCF = 6.7 Hz), 26.1. 19F NMR (282 MHz, CDCl3) δ − 228.0 (td, JHF = 47.1, 17.5 Hz). HRMS-MALDI (M + Na)+: Calculated for [C14H16FNO3+Na]+: 288.1012; Found: 288.0995.
General procedure for the amide coupling to synthesize 6a–i
To intermediate 1 (150 mg, 0.565 mmol) were added 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) (217.7 mg, 0.678 mmol), acetonitrile (15 mL) and triethylamine (TEA) (197 uL, 1.413 mmol). The mixture was stirred at rt for 0.5 h. The nucleophile (amine, alcohol or amino acid) (1.2–2 equiv.) was added. The resulting mixture was stirred at rt for 2 h (6c), or over one (6a) to two nights (6e, 6f), or at 65 °C for 2–6 h (6d, 6g–h) or over five nights (6i). The reaction mixture was concentrated, then partitioned in EtOAc/water (15 mL/15 mL). For 6g–h, the water phase was acidified using 10% KHSO4 aqueous solution to pH at about 5. After further extraction with EtOAc (2 × 15 mL), concentration and purification using silica gel or preparative LC, the final products 6a–6i were achieved in 56–91% yield. All the products were analyzed using LC and showed more than 96% purity. LC condition: B:A (10:90–80:20, 10 mM NH4OAc, gradient time: 4 min, hold time: 2 min. X-Bridge C18, 3.5 um. Diastereoisomers 6e–h showed as one peak under this condition (Appendix A of the Supplemental part).
Synthesis of N-((3s,5s,7s)-adamantan-1-yl)-1-(5-fluoro-4-hydroxypentyl)-1H-indole-3-carboxamide (6a)
Following the general procedure, adamantine hydrochloride (127.3 mg, 0.678 mmol) was used, and the reaction mixture was stirred at rt overnight. The crude product was purified using silica gel with EtOAc/n-heptane (50:50–70:30) to give the product 6a (143.5 mg, 64%) as a while solid. 1H-NMR (300 MHz, CDCl3): δ 7.90–7.83 (m, 1H), 7.64 (s, 1H), 7.36–7.30 (m, 1H), 7.25–7.19 (m, 2H), 5.75 (br s, 1H), 4.40–4.14 (m, 2H), 4.14–4.08 (m, 2H), 3.80 (m, 1H), 2.19–2.10 (m, 9H), 2.07–1.85 (m, 2H), 1.81–1.67 (m, 6H), 1.46–1.36 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 164.7, 136.6, 131.4, 125.3, 122.4, 121.3, 120.0, 112.3, 110.4, 86.8 (d, JCF = 170 Hz), 69.8 (d, JCF = 19.4 Hz), 52.2, 46.5, 42.3, 36.6, 29.7, 29.1 (d, JCF = 5.9 Hz), 26.1. 19F-NMR (282 MHz, CDCl3): δ − 227.55 (td, JHF = 47.5, 17.4 Hz). HRMS-MALDI (M + H)+: Calculated for [C24H31FN2O2+H]+: 399.2448; Found: 399.2445.
Conclusion
A straightforward synthetic route was developed for the synthesis of a key intermediate on a multigram scale to be used for the synthesis of potential synthetic cannabinoid metabolites with a 5F-4OH pentyl side chain. The synthetic route, which could be potentially used in industrial production, was applied in the synthesis of nine potential metabolites of eight different synthetic cannabinoids. The key intermediate could be used as a product to meet the different needs in different labs for synthesis of potential synthetic cannabinoid metabolites with a 5F-4OH pentyl side chain. Similar synthesis strategies could be applied in the synthesis of similar metabolites of other synthetic cannabinoids with different lengths of fluorinated side chains for example 6-fluoro hexyl, or with a different core, such as indazole.
Supplemental Material
Download MS Word (10.6 MB)Additional information
Funding
References
- United Nations Office on Drugs and Crime (UNODC) 2020 Current NPS Threats, Volume II, January 2020. https://www.unodc.org/documents/scientific/Current_NPS_Threats_Volume_II_web.pdf.
- Uchiyama, N.; Asakawa, K.; Kikura-Hanajiri, R.; Tsutsumi, T.; Hakamatsuka, T. Forensic Toxicol. 2015, 33, 367–373. DOI: 10.1007/s11419-015-0283-8.[10.1007/s11419-015-0268-7]
- Mississippi State Department of Health. Mississippi Morb. Rep. 2015, 31, 1–5. https://msdh.ms.gov/msdhsite/_static/resources/6307.pdf.
- Grigoryev, A.; Melnik, A.; Savchuk, S.; Simonov, A.; Rozhanets, V. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 2519–2526. DOI: 10.1016/j.jchromb.2011.07.004.
- Cannaert, A.; Storme, J.; Franz, F.; Auwärter, V.; Stove, C. P. Anal. Chem. 2016, 88, 11476–11485. DOI: 10.1021/acs.analchem.6b02600.
- Carlier, J.; Diao, X.; Wohlfarth, A.; Scheidweiler, K.; Huestis, M. A. Curr. Neuropharmacol. 2017, 15, 682–691. DOI: 10.2174/1570159X15666161108123419.
- Castaneto, M. S.; Scheidweiler, K. B.; Gandhi, A.; Wohlfarth, A.; Klette, K. L.; Martin, T. M.; Huestis, M. A. Drug Test Anal. 2015, 7, 483–493. DOI: 10.1002/dta.1709.
- Diao, X.; Huestis, M. A. Front. Chem. 2019, 7, 109. DOI: 10.3389/fchem.2019.00109.
- Vikingsson, S.; Josefsson, M.; Gréen, H. J. Anal. Toxicol. 2015, 39, 426–435. DOI: doi.org/10.1093/jat/bkv045. DOI: 10.1093/jat/bkv045.
- Wallgren, J.; Vikingsson, S.; Åstrand, A.; Josefsson, M.; Gréen, H.; Dahlén, J.; Wu, X.; Konradsson, P. Tetrahedron 2018, 74, 2905–2913. DOI: 10.1016/j.tet.2018.04.026.
- Wallgren, J.; Vikingsson, S.; Johansson, A.; Josefsson, M.; Green, H.; Dahlén, J.; Wu, X.; Konradsson, P. Tetrahedron Lett. 2017, 58, 1456–1458. DOI: 10.1016/j.tetlet.2017.02.077.
- Longworth, M.; Connor, M.; Banister, S. D.; Kassiou, M. ACS Chem. Neurosci. 2017, 8, 1673–1680. DOI: 10.1021/acschemneuro.7b00116.
- Gamage, T. F.; Farquhar, C. E.; McKinnie, R. J.; Kevin, R. C.; McGregor, I. S.; Trudell, M. L.; Wiley, J. L.; Thomas, B. F. J. Pharm. Exp. Ther. 2019, 368, 411–422. DOI: 10.1093/scan/nsz022.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). Thematic Papers: Understanding the ‘Spice’ Phenomenon. 2009. DOI: 10.2810/27063.
- Schoeder, C. T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C. E. Forensic Toxicol. 2018, 36, 385–403. DOI: 10.1007/s11419-018-0415-z.
- Angerer, V.; Franz, F.; Moosmann, B.; Bisel, P.; Auwärter, V. Forensic Toxicol. 2019, 37, 186–196. DOI: 10.1007/s11419-018-0451-8.
- Patton, A. L.; Seely, K. A.; Chimalakonda, K. C.; Tran, J. P.; Trass, M.; Miranda, A.; Fantegrossi, W. E.; Kennedy, P. D.; Dobrowolski, P.; Radominska-Pandya, A.; et al. Anal. Chem. 2013, 85, 9390–9399., DOI: 10.1021/ac4024704.
- McKinnie, R. J.; Darweesh, T.; Zito, P. A.; Shields, J.; Trudell, M. L. Synthesis 2018, 50, 4683–4689. DOI: 10.1055/s-0037-1609914.
- Wallgren, J.; Rexander, A.; Vestling, E.; Liu, H.; Dahlén, J.; Konradsson, P.; Wu, X. Synlett 2020, 31, 517–520. DOI: 10.1055/s-0039-1691571.
- Hess, C.; Schoeder, C. T.; Pillaiyar, T.; Madea, B.; Müller, C. E. Forensic Toxicol. 2016, 34, 329–343. DOI: 10.1007/s11419-016-0320-2.
- Banister, S. D.; Longworth, M.; Kevin, R.; Sachdev, S.; Santiago, M.; Stuart, J.; Mack, J. B. C.; Glass, M.; McGregor, I. S.; Connor, M.; Kassiou, M. ACS Chem. Neurosci. 2016, 7, 1241–1254. DOI: 10.1021/acschemneuro.6b00137.
- Longworth, M.; Banister, S. D.; Boyd, R.; Kevin, R. C.; Connor, M.; McGregor, I. S.; Kassiou, M. ACS Chem. Neurosci. 2017, 8, 2159–2167. DOI: 10.1021/acschemneuro.7b00267.