
Abstract
Background: Very few mitochondrial myopathies have been described in horses.
Objective: To examine the ultrastructure of muscle mitochondria in equine cases of myopathy of unknown origin.
Materials & methods: Biopsies of vastus lateralis of the Musculus quadriceps femoris were taken predominantly immediately post mortem and processed for transmission electron microscopy. As a result, electron micrographs of 90 horses in total were available for analysis comprising 4 control horses, 16 horses suffering from myopathy and 70 otherwise diseased horses.
Results: Following a thorough clinical and laboratory work-up, four out of five patients that did not fit into the usual algorithm to detect known causes of myopathy showed ultrastructural mitochondrial alterations. Small mitochondria with zones with complete disruption of cristae associated with lactic acidemia were detected in a 17-year-old pony mare, extremely long and slender mitochondria with longitudinal cristae in a 5-year-old Quarter horse stallion, a mixture of irregular extremely large mitochondria (measuring 2500 by 800 nm) next to smaller ones in an 8-year-old Hanoverian mare and round mitochondria with only few cristae in a 11-year-old pony gelding. It remains uncertain whether the subsarcolemmal mitochondrial accumulations observed in the fifth patient have any pathological significance.
Conclusions: Ultrastructural alterations in mitochondria were detected in at least four horses. To conclude that these are due to mitochondrial dysfuntions, biochemical tests should be performed.
Practical applications: The possibility of a mitochondrial myopathy should be included in the differential diagnosis of muscle weakness.
1. Introduction
Muscle weakness in horses can have many different causes. It can be a sign of various systemic abnormalities, but can also be due to a disease that primary causes muscle damage, called myopathy (Aleman Citation2008). Following a thorough clinical work-up, the vast majority of patients can be classified in one of the categories of known myopathies. This is definitely helpful, even if for several of these categories, such as atypical myopathy (also known as equine acquired multiple acyl-CoA dehydrogenase deficiency (MADD)), the therapeutic possibilities are limited (Van Galen et al. Citation2012). A number of patients, however, cannot be classified into the known categories of equine myopathy. Some of these patients have long-lasting complaints, possibly pointing to a congenital/hereditary defect. (Encephalo)myopathies in humans can be associated with mutations in mitochondrial genes or nuclear genes that encode for mitochondrial proteins (Dimauro & Davidzon Citation2005). Mitochondrial ultrastructural alterations are extensively described in human mitochondrial myopathies (Haas et al. Citation2008). The replication of the mitochondrial genome is not linked to the cell cycle. Mutations in mitochondrial DNA (mtDNA) can coexist with wild-type mtDNA in a somatic cell which is called heteroplasmy (Dimauro & Davidzon Citation2005). There is a continuous replication of mtDNA, which is thought to be more susceptible to mutations than nuclear DNA (Brown et al. Citation1979). During cell division, a daughter cell can undergo a shift in the proportion of mutant mtDNA, which can alter the phenotype by exceeding a certain threshold (Dimauro & Davidzon Citation2005). This could explain why mitochondrial diseases can arise at any age. Mitochondrial disorders are not as rare as previously assumed. In fact, they are amongst the most common inherited diseases in man (Schaefer et al. Citation2004). There is no reason to believe that this should be different in animals. Nevertheless, little is known about mitochondrial myopathies in domestic animals in general and in horses in particular. Very few patients with mitochondrial myopathy have been described in the veterinary literature. In fact, in horses, to our knowledge only one case report describes a Complex I deficiency in an Arabian mare with marked lactic acidosis (Valberg et al. Citation1994). In this patient, large accumulations of mitochondria with bizarre cristae have been observed by transmission electron microscopy. Other reports of ultrastructural alterations in mitochondria include the changes seen in horses with acquired MADD (Cassart et al. Citation2007) and the subsarcolemmal accumulation of swollen mitochondria in horses with pituitary pars intermedia dysfunction myopathy (Aleman et al. Citation2006).
The purpose of this article was to examine mitochondrial ultrastructure in equine cases of myopathy of unknown origin. Here, we report the aberrant mitochondrial structure in five equine patients of various breed and age, diagnosed with myopathy of unknown origin.
2. Materials and methods
2.1. Animals
Biopsies of vastus lateralis of the Musculus quadriceps femoris were taken predominantly immediately post mortem and processed for transmission electron microscopy. As a result, electron micrographs of 90 horses in total were available for analysis comprising 4 control horses, 16 horses suffering from myopathy and 70 otherwise diseased horses. Emphasis was laid on five horses with myopathy of unknown origin in this article, because of their remarkable ultrastructural changes: patient 1, a 17-year-old pony mare; patient 2, a five-year-old Quarter horse stallion; patient 3, an eight-year-old Hanoverian mare; patient 4, an 11-year-old pony gelding; and patient 5, a two-year-old Dutch Warmblood colt.
2.2. Clinical examination
All horses were admitted to the Equine Clinic, Faculty of Veterinary Medicine, Utrecht University, the Netherlands. Routine clinical examination as well as blood and urine examination were performed. Further examinations were performed, depending on the results of the first tests. Electromyography (EMG) was performed for patients 2, 3 and 4. Organic acid profiling was performed for patients 2, 3 and 4 and acylcarnitine profiling (AP) for patients 1, 3 and 4. A genetic test for hyperkalemic periodic paralysis (HYPP) was done for patient 2. Patient 3 underwent radiological examination to exclude orthopedic problems.
2.3. Biopsy
A Bergström needle (7 mm in diameter) was introduced in the vastus lateralis of the Musculus quadriceps femoris. The muscle biopies were performed in vivo for patients 1, 2 and 3 and within fifteen minutes after euthanasia for the two remaining horses.
2.4. Sample preparation for light and electron microscopy
The samples for histochemistry were snap frozen in liquid nitrogen preceded by pre-cooling in isopentane. Transverse sections were cut with a cryostat and stained with haematoxylin and eosin, periodic acid Schiff, Sudan Black B, ATPase (pH 4.3 and 9.4), NADH-TR, succinate dehydrogenase (SDH), cytochrome c-oxidase (COX) and modified Gomori trichrome stains for light microscopy. COX and SDH enzyme histochemistry was performed only for patients 1, 2 and 3.
Samples for transmission electron microscopy were fixed in paraformaldehyde (1%) and glutaraldehyde (2.5%) in cacodylate buffer, and embedded in epoxy resin medium. Ultrathin sections were made and contrasted with uranyl acetate and lead citrate.
3. Results
3.1. Case history and clinical examination
The case history data are summarized in .
Table 1. The onset of the disease, the clinical signs, treatment and outcome of five cases of equine myopathy of unknown origin.
Patient 1 was presented with intention tremor and hypermetria of the hindquarters since less than one day. She recovered from clinical signs and was still alive after a follow-up period of five years. Patient 2 had suffered from weakness in the hind limbs and weight loss for several weeks. He recovered from the clinical signs and was still alive after a follow-up period of five years. Patient 3 had suffered from progressive weight loss and muscle atrophy for several months. She also recovered and was still alive after four months of follow-up.
Patient 4 was euthanized after suffering several months from a myopathy with atrophy of the back muscles and exercise intolerance. Patient 5 died within a few hours after presentation in the clinic with exercise intolerance, muscle tremor and heavy sweating.
OAP and AP tests were within reference values. The test for HYPP in patient 2 was negative. Radiologic examination in patient 3 revealed a mild arthrosis in two cervical joints (C5/C6 and C6/C7). According to EMG, patients 2 and 3 were diagnosed with myopathy and the analyses in patient 4 fitted with a diagnosis of muscle atrophy.
3.2. Blood and urine examination
Results of the standard blood and urine analyses are summarized in . Most remarkable in patient 1 was the severe lactic acidosis in peripheral blood.
Table 2. Urine and blood analyses in five horses with myopathy of unknown origin.
3.3. Light microscopy
Histological abnormalities in muscle were detected in patients 4 and 5. Patient 4 showed several muscle fibers with degeneration. Patient 5 showed multifocal individually shrunken, swollen or fragmented fibers, strongly eosinophilic, with hyalinization, loss of striation and pyknotic nuclei. No abnormalities were detected with the other staining techniques.
3.4. Ultrastructure
In none of the five patients, the typical sarcoplasmic lipid droplets and swollen mitochondria with cristae fragmentation, as described in equine atypical myopathy (Cassart et al. Citation2007), were observed under the electron microscope.
In patient 1, ultrastructural examination of the vastus lateralis muscle revealed the presence of small, round to oval shaped intermyofibrillar mitochondria, the longest diameter of which was less than 500 nm ((a)). Their cristae appeared to be fewer in number, swollen and less complex ((b)). Some mitochondria had brighter zones (arrow) and zones with complete disruption of the cristae (asterisk).
Figure 1. Transmission electron microscopy of the vastus lateralis muscle in patient 1, a 17-year-old pony mare: small round to oval mitochondria line up between the myofibrils (a, arrows); scale 1a = 2 μm. Small mitochondria with few, simple cristae were seen, some presenting with brighter zones in the mitochondrial matrix (b, arrow) and zones with complete disruption of cristae (b, asterisk); scale 1b = 500 nm.
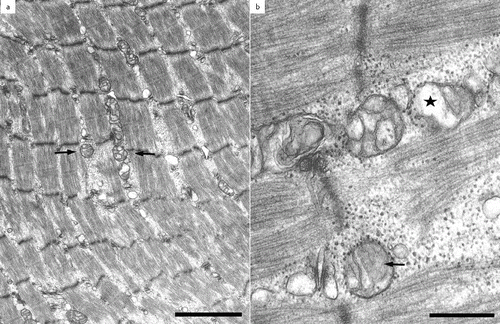
Transmission electron microscopy in patient 2 revealed extremely long and slender mitochondria with longitudinal cristae ( (a)). Many of the mitochondria were more than 2000 nm in length but less than 150 nm in width. On transverse section, the cristae gave the mitochondria an onion-shaped appearance as they formed concentric lamellae ((b)).
Figure 2. Transmission electron microscopy of the vastus lateralis muscle in patient 2, a five-year-old Quarter horse stallion: note extremely long and slender mitochondria with longitudinal cristae (a). Some mitochondria show an onion-shaped aspect on transverse section due to cristae forming concentric lamellae (b).
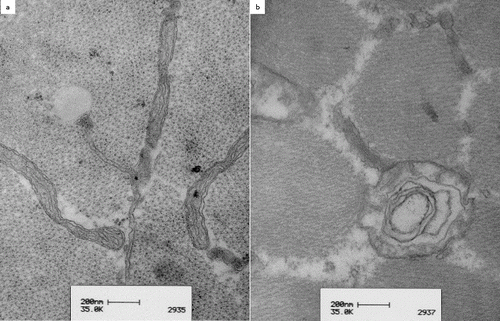
In patient 3, ultrastructural examination of the vastus lateralis muscle revealed a mixture of irregular extremely large mitochondria, measuring 2500 by 800 nm ((a), large arrow) next to smaller ones ((a), small arrows). These large mitochondria were present both in between the myofibrils and under the sarcolemma. Branching cristae were seen in the large mitochondria ((b)).
Figure 3. Transmission electron microscopy of the vastus lateralis muscle in patient 3, an eight-year-old Hanoverian mare: simultaneous presence of mitochondria of normal size and shape (a, small arrows) and large mitochondria (a, thick arrow) were seen. The right picture shows a detailed view of an extremely large mitochondrion with complex branching cristae (b).
In patient 4, electron microscopy showed round mitochondria of approximately 150 to 400 nm in diameter, with only few cristae ((a)).
Figure 4. Transmission electron microscopy of the vastus lateralis muscle in patient 4, an 11-year-old pony gelding: small round mitochondria with only few cristae were seen (a). Transmission electron microscopy of the vastus lateralis muscle in patient 5, a two-year-old Dutch Warmblood colt: subsarcolemmal accumulations of mitochondria were present (b).
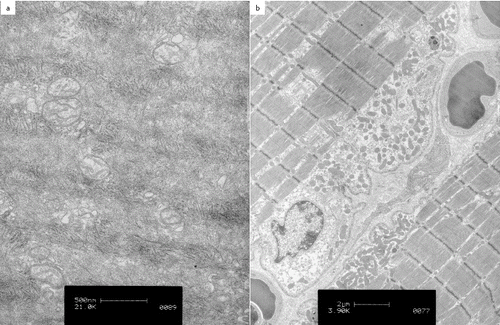
Electron microscopy of the vastus lateralis muscle in patient 5 revealed subsarcolemmal accumulations of mitochondria ((b)).
4. Discussion
Although in recent years, there is some interest in mitochondrial phenotyping and genotyping in horses, especially with respect to racing performance (Harrison & Turrion-Gomez Citation2006), and although the equine mitochondrial genome has been sequenced (Jiang et al. Citation2011), still very little is known about pathological mitochondrial structure and function in horses.
Mitochondria have long been believed to be a heterogeneous group of individual bean-shaped isolated organelles randomly distributed within the cytoplasm. These organelles are now known to form dynamic tubular networks constantly evolving through processes of fission and fusion (Okamoto & Shaw Citation2005). Under physiological circumstances, the frequencies of these two processes are balanced so that the overall morphology of the mitochondrial population is maintained (Detmer & Chan Citation2007). These processes also have an important impact on the function of mitochondria and dysfunctions in these processes can have important consequences for development, disease and apoptosis (Detmer & Chan Citation2007). Therefore, mitochondrial fusion and fission are two strictly controlled processes, involving the coordination of four membranes. Three large GTP-ases, OPA1 and the mitofusins Mfn1 and Mfn2, are essential for the fusion of mammalian mitochondria (Song et al. Citation2009). Although most studies concerning mitochondrial dynamics have been limited to yeast, cultured mammalian cells and neurons, it has recently been shown that mitochondrial fusion in skeletal muscle is important for the protection of mtDNA (Chen et al. Citation2010). Mice lacking mitofusin function in skeletal muscle develop a lethal mitochondrial myopathy. Mitofusin is required for fusion of the outer mitochondrial membranes (Koshiba et al. Citation2004), as OPA1 is proven to be necessary for the inner mitochondrial membrane fusion (Song et al. Citation2009). Unlike in yeast cells, the fusion of the outer and inner membranes does not seem to be as tightly coupled in mammalian cells. A dysfunction in mitochondrial fusion can lead to small or fragmented mitochondria. It is possible that patient 1 is an example of a dysfunction of mitochondrial fusion. In human medicine, lactic acidosis, as reported in this patient, is associated with mitochondrial dysfuntions (De Vivo & Dimauro Citation1990).
Mitochondrial fission is a process controlled by the interaction of dynamin-related protein 1 (Drp1) and fission protein 1 (Fis1) (Westermann Citation2010). A decrease in fission can lead to elongated, interconnected mitochondria, as seen in patient 2. Elongated mitochondria are also reported during autophagy (Gomes et al. Citation2011). In patient 2, however, autophagy was not observed.
Large mitochondria with complex cristae were observed in the vastus lateralis muscle of patient number 3. Giant mitochondria with complex cristae conformation have been occasionally reported in the myocardium of human patients with mitochondrial cardiomyopathy (Kanzaki et al. Citation2010). In our equine patient, however, there were no clinical indications of a cardiomyopathy. Recently, human patients were described concerning patients suffering from congenital megaconial myopathy due to a defect in the choline kinase beta gene. It is thought that an impaired phospholipid metabolism in the mitochondria-associated membrane causes abnormal size and displacement of the mitochondria to the periphery of the fibers. On COX, SDH and modified Gomori trichrome stains, these mitochondria can be seen as large dots (Mitsuhashi & Nishino Citation2013). Histology in patient 3, however, did not match megaconial myopathy.
Swelling of mitochondria can be a manifestation of hyperosmotic stress (Cheville Citation1994). The cristae, however, in patient 4 had a normal appearance.
Finally, in patient 5, the only ultrastructural alteration observed was the accumulation of mitochondria under the sarcolemma. This was, however, not noticed as ragged red fibers at light microscopic level, as it is usually reported in humans (Rifai et al. Citation1995). Subsarcolemmal mitochondrial proliferations have been described in skeletal muscle from human patients with a defect in mtDNA (Bourgeois & Tarnopolsky Citation2004). These accumulations have, however, also been reported to increase with normal ageing and may reflect a mere age-related decline in muscle mitochondrial oxidative metabolism (Rifai et al. Citation1995). Patient 5 in our series, however, was only two years old.
In conclusion, ultrastructural alterations in mitochondria in equine myopathy of unknown origin were detected in five patients. Aberrant mitochondrial ultrastructure is repeatedly described and discussed in human medical literature. However, this is not specific for mitochondrial myopathy and can be due to other disorders (Haas et al. Citation2008), which raises the question of whether the ultrastructural changes are associated with functional abnormalities and account for the clinical signs of myopathy. This points out the need for a standardized diagnostic protocol. In order to progress in our understanding of equine mitochondrial myopathies, future diagnostic approach should include analyses of muscle biopsies with histochemical functional assays, electron microscopy and respiratory chain enzyme assays, as proposed for humans (Pfeffer & Chinnery Citation2013). Based on these tests and on the clinical features and the inheritance pattern of the suspected mitochondrial myopathy, genetic tests can be performed (Pfeffer & Chinnery Citation2013). Specific histochemical enzyme reactions used for the diagnosis of mitochondrial diseases are the SDH and the COX reactions (Taylor et al. Citation2004). Activities of mitochondrial complexes have been determined in Dutch Warmblood mares (Westermann et al. Citation2011). This gives the possibility to reveal equine mitochondrial myopathies in the future.
Acknowledgements
The technical support of Ton Ultee is greatly appreciated.
This article was originally published with errors. This version has been corrected. Please see Corrigendum (http://dx.doi.org/10.1080/01652176.2015.1008273).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Aleman M. 2008. A review of equine muscle disorders. Neuromuscul Disord. 18:277–287.
- Aleman M, Watson JL, Williams DC, LeCouteur RA, Nieto JE, Shelton GD. 2006. Myopathy in horses with pars intermedia dysfunction (Cushing's disease). Neuromuscul Disord. 16:737–744.
- Bourgeois JM, Tarnopolsky MA. 2004. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion. 4:441–452.
- Brown WM, George M Jr, Wilson AC. 1979. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA. 76:1967–1971.
- Cassart D, Baise E, Cherel Y, Delguste C, Antoine N, Votion D, Amory H, Rollin F, Linden A, Desmecht D. 2007. Morphological alterations in oxidative muscles and mitochondrial structure associated with equine atypical myopathy. Equine Vet J. 39:26–32.
- Chen H, Vermulst M, Wang Y, Chomyn A, Prolla T, McCaffery J, Chan D. 2010. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 141:280–289.
- Cheville FN. 1994. Ultrastructural pathology: an introduction to interpretation. Ames (IA): Iowa State University Press. Chapter 2, Interpretation of acute cell injury: degeneration; p. 51–76.
- Detmer S, Chan D. 2007. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 8:870–879.
- De Vivo DC, Dimauro S. 1990. Mitochondrial defects of brain and muscle. Biol Neonate. 58:54–69.
- Dimauro S, Davidzon G. 2005. Mitochondrial DNA and disease. Ann Med. 37:222–232.
- Gomes LC, Di Benedetto G, Scorrano L. 2011. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 13:589–598.
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Wong LJ, Cohen BH, Naviaux RK. 2008. The in-depth evaluation of suspected mitochondrial disease. Mol Gen Metab. 1:16–37.
- Harrison S, Turrion-Gomez J. 2006. Mitochondrial DNA: an important female contribution to thoroughbred racehorse performance. Mitochondrion 6:53–66.
- Jiang Q, Wei Y, Huang Y, Jiang H, Guo Y, Lan G, Liao, DJ. 2011. The complete mitochondrial genome and phylogenetic analysis of the Debao pony (Equus Caballus). Mol Biol Rep. 38:593–599.
- Kanzaki Y, Terasaki F, Okabe M, Otsuka K, Katashima T, Fujita S, Takahide I, Kitaura Y. 2010. Giant mitochondria in the myocardium of a patient with mitochondrial cardiomyopathy, transmission and 3-dimensional scanning electron microscopy. Circulation. 121:831–832.
- Koshiba T, Detmer S, Kaiser J, Chen H, McCaffery J, Chan D. 2004. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 305:858–862.
- Mitsuhashi S, Nishino I. 2013. Megaconial congenital muscular dystrophy due to loss-of-function mutations in choline kinase beta. Curr Opin Neurol. 26:536–543.
- Okamoto K, Shaw J. 2005. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Ann Rev Genet. 39:503–536.
- Pfeffer G, Chinnery PF. 2013. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 45:4–16.
- Rifai Z, Welle S, Kamp C, Thornton CA. 1995. Ragged red fibers in normal aging and inflammatory myopathy. Ann Neurol. 37:24–29.
- Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. 2004. The epidemiology of mitochondrial disorders-past, present and future. Biochim Biophys Acta. 1659:115–120.
- Son Z, Ghochani M, McCaffery J, Frey T, Chan D. 2009. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Molec Biol Cell. 20:3525–3532.
- Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. 2004. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord. 14:237–245.
- Valberg SJ, Carlson GP, Cardinet GH, Birks EK, Jones JH, Chomyn A, DiMauro S. 1994. Skeletal muscle mitochondrial myopathy as a cause of exercise intolerance in a horse. Muscle Nerve. 17:305–312.
- Van Galen G, Saegerman C, Pitel CM, Patarin F, Amory H, Baily JD, Cassart D, Gerber V, Hahn C, Harris P, et al. 2012. European outbreaks of atypical myopathy in grazing horses (2006–2009): determination of indicators for risk and prognostic factors. Equine Vet J. 44:621–625.
- Westermann B. 2010. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 11:872–884.
- Westermann CM, Dorland L, van Diggelen OP, Schoonderwoerd K, Bierau J, Waterham HR, van der Kolk JH. 2011. Decreased oxidative phosphorylation and PGAM deficiency in horses suffering from atypical myopathy associated with acquired MADD. Mol Genet Metab. 104:273–278.