
Abstracts
Purpose
High-throughput sequencing technologies have revealed that the lungs contain a variety of low biomass microbiota associated with various lung diseases. Rat model is an important tool to understand the possible causal relationship between pulmonary microbiota and diseases. Antibiotic exposure can alter the microbiota, however, a direct influence of long-term ampicillin exposure on commensal bacteria of healthy lungs has not been investigated, which could be useful in the study of the relation between microbiome and long-term lung diseases, especially in animal model-making of lung diseases.
Methods
The rats were aerosolized ampicillin of different concentrations for five months, and then the effect on the lung microbiota was investigated using 16S rRNA gene sequencing.
Results
The ampicillin treatment by a certain concentration (LA5, 0.2 ml of 5 mg/ml ampicillin) administration leads to profound changes in the rat lung microbiota but not in the low critical ampicillin concentration (LA01 and LA1, 0.1 and 1 mg/ml ampicillin), when compared to the untreated group (LC). The genus Acidobacteria_Gp16 dominated the ampicillin treated lung microbiota while the genera Brucella, Acinetobacter, Acidobacteria_Gp14, Sphingomonas, and Tumebacillus dominated the untreated lung microbiota. The predicted KEGG pathway analysis profile revealed some difference in the ampicillin treated group.
Conclusions
The study demonstrated the effects of different concentrations of ampicillin treatment on lung microbiota of rats in a relatively long term. It could serve as a basis for the clinical use of antibiotic and the use of ampicillin to control certain bacteria in the animal model-making of respiratory diseases such as chronic obstructive pulmonary disease.
Introduction
The host cell and symbiotic microorganisms form a superorganism in the human body, which collectively determines the disease and health of the body. The dysbiosis of symbiotic microbiota can modulate the progression of human disease or cause the disease.Citation1 The lungs are traditionally considered to be sterile, however, with the technological progress of high-throughput sequencing and bioinformatics analysis, a dynamic and complex microbial community has been found in the lung mucosa in both healthy and disease states.Citation2–5 It has been found that the lung bacterial community structure could respond to diseases such as asthma,Citation3 infections by pathogens,Citation6,Citation7 cystic fibrosis,Citation8 chronic obstructive pulmonary disease (COPD),Citation9 lung injury,Citation10 or lung transplantation.Citation11
Disturbance of the microbial diversity and stability has been shown to correlate with immunologic, diverse metabolic, and inflammatory disorders.Citation12 Antibiotic exposure can alter the microbiota and is linked to the development and progression of disease. Most of the microbiome studies have focused on the gut microbiome and have examined the effects of various antibiotics on gut microbiota,Citation13–16 yet lung microbiome study is still in its infancy and facing a series of critical challenges.Citation17 Aerosolization and nasal sprays are efficient and noninvasive methods to deliver molecules such as antibiotics to the specific body sites.Citation18 In humans, inhaled antibiotics have been used to treat critical lung infections,Citation19 and nasal spray antibiotics have been reported to prevent acute otitis caused by Streptococcus salivarius in children.Citation20 And therefore they represent a strategy to locally and efficiently modify the lung microbiota that could limit the exposure of other organs in the body. Ampicillin is the common clinical antibiotic used to treat a variety of bacterial infections. Indications include respiratory infections, urinary tract infections, meningitis, salmonellosis, and endocarditis. It can also be used to prevent group B streptococcal infection in newborns.
COPD is a common and frequently occurring disease of respiratory system, characterized by persistent respiratory symptoms and airflow limitation is one of the leading causes of morbidity and mortality worldwide.Citation21,Citation22 The relationship between the changes in healthy lung microbiota and the progression of early COPD remains unclear. Antibiotic exposure can alter the microbiota. And a number of studies have found that long-term prophylactic antibiotic use in COPD for a period of up to 6 or 12 months, could reduce the number of patients with exacerbations and exacerbation frequency, and increase possibly health-related quality of life.Citation23,Citation24 Animal models are valuable tools to investigate the lung microbiota as technical and ethical restrictions often limited the direct lung studies in humans. The rat is the most commonly used animal model. The COPD rat model-making needed to be exposed to smoke for 20 week.Citation25 During the time of the 20 week of model-making, using the antibiotic to intervene in the lung microbiota could benefit to uncover the relationship between the lung microbiota change in healthy rats and the formation of COPD. The maximal duration of continuous prophylactic antibiotics was 12 months, and for pulsed antibiotics it was 36 months.Citation23,Citation24 There are no data on the impact of very long-term antibiotic use on the lung microbiota of healthy rats.
Herein, as the lung diseases such as the chronic obstructive pulmonary disease (COPD) were the long-term disease, we expected to study the relationship between lung microbial community change and the lung disease, by long-term interference using antibiotic. We investigated the effect of long-term and different concentrations of low doses of ampicillin on the pulmonary microbiota in a healthy rat model in the present study. We hypothesized that a long duration of antibiotic may lead to substantial changes in the pulmonary microbiota and the different concentrations of ampicillin may shape different lung microbiota. It could provide basis for further research on the relationship between the changes in bacterial flora structure and the occurrence and development of early COPD, and also provide some reference for clinically rational long-term prophylactic antibiotic use.
Materials and methods
Rats and sample collection
Male specific-pathogen-free (SPF) Sprague-Dawley (SD) rats (6 week in age) weighing 200 ± 20 g were obtained from Guangdong Medical Laboratory Animal Center (Guangdong, China) Biotechnology Co., Ltd. (Beijing, China). The animal care and experiments in this research were in accordance with the guiding principles for the care and use of laboratory animals recommended by the Chinese Association for Laboratory Animal Science Policy. It was approved by the Institutional Animal Care and Use Committee at the Guangzhou Medical University of China (Guangzhou, China; No. 2019-159). The rats were kept in a specific pathogen-free (SPF) room on alternate 12-h light/dark cycles at maintained temperature control and 50% humidity. They had free access to standardized food and water.
All rats were allowed to adjust to their environment for one month in four-animal cages in a common animal housing room on the free-standing shelves in the same one cabinet sharing the ventilation system before the experiments. To account for cage effect,Citation25,Citation26 rats were assigned randomly into four groups: LC (n = 4), LA01 (n = 8), LA1 (n = 8), and LA5 (n = 8). Considering the previous microbiome-related studies by using the animal models and the complicated long-term antibiotic treatment with different concentrations which was difficult to accomplish for a big sample size, the number of 8 consistent genetic background rats were set for each group to explore effect of ampicillin antibiotic on lung microbiome. The LA01, LA1, and LA5 groups were administered aerosolized of 0.2 ml ampicillin with the concentration of 0.1 mg/ml (LA01), 1 mg/ml (LA1), and 5 mg/ml (LA5) once daily for one month. Then the antibiotic doses were adjusted to every other day until six months. The dosage utilized was determined according to the other similar researches about antibiotic disturbance in animal models.Citation27–30 The control group of LC did not receive aerosolization of ampicillin. The aerosolization of ampicillin was performed by the same experienced researcher using Micro Sprayer Aerosolizer (YUYAN Instrument, Shanghai, China), which was the standard aerosolization equipment.Citation31 In order to avoid the continuous blockage of the rats’ noses and mouths by using isoflurane combined with a small animal anesthesia machine,Citation32,Citation33 the rats were anesthetized with isoflurane by putting the cotton balls with 1–2 ml of isoflurane in the rat cage for 1–2 min. As described previously, the rats were held supine on a 60° incline board by having their upper incisor teeth after anesthesia. The mouth of the rats was gently opened due to the weight of the rats, and the rats were intubated through the mouth, and trachea using Micro Sprayer Aerosolizer inserted 10 mm into the trachea with a bulb-headed laryngoscope.Citation31 We then used the nebulizing needle to aerosolize ampicillin into the airway through the epiglottis. After the treatment every time, the rats were housed in three or four-animal cages in sequential weeks over a 20 week period in the same room on free-standing shelves in the same one cabinet sharing the ventilation system.
After the six months, rats were then sacrificed under sodium pentobarbital anesthesia by neck removal to minimize suffering for lung tissue collection. The lung tissues of each rat were collected into 15 ml centrifuge tubs with 5 mL phosphate-buffered saline (PBS), immediately placed in liquid nitrogen, and stored at −80 °C for future analysis. The surgical scissors were cleaned and rinsed in ethanol, PBS and flamed between each organ. PBS control specimens from surgical scissors rinsed with clean instruments were included as procedural negative controls for whole lung tissue. The PBS on the sampling bench during the whole working time and blank DNA extraction without any sample template served as negative controls.
Lung microbial DNA extraction and 16S rRNA gene sequencing
Isolating the total DNA from lung tissue was accomplished using the Qiagen genomic DNA isolation kit according to the manufacturer’s instructions. The V3-V4 regions of the 16S rDNA were amplified using the primers 341 F (5′-CCTACGGGNGGCWGCAG-3′) and 806 R (5′-GACTACHVGGGTATCTAATCC-3′) combined with the adapter and barcode sequences. All PCR reactions were carried out in 10 μL reactions with 5 μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.3 μM of forward and reverse primers, and about 50 ng template DNA. The reaction started with initialization at 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 30 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 40 s. And the reaction was completed with a final elongation step at 72 °C for 7 min. Three technical replicates for every sample were prepared and pooled during purification with OMEGA DNA purification Kit and Monarch DNA Gel Extraction Kit. Purified PCR products were quantified using Qubit®3.0 (Life Invitrogen) and mixed equally. The sequencing of pooled 16S rRNA gene was performed on an Illumina NovaSeq 6000 platform using the 250 paired-end protocol at Biomarker Technologies Corporation (Beijing, China). Sequence data is available at the ENA Accession number: PRJNA862086.
Bioinformatic and statistical analysis
Raw data were demultiplexed into individual samples according to the unique barcodes and then quality controlled using FastQC.Citation34 Forward and reverse end sequences of each sample were merged using Vsearch v2.21.1.Citation35 The sequences and the chimeric sequences were removed by Vsearch v2.21.1. Then the high-quality sequence analysis was performed by QIIME2 pipeline.Citation36 The Unoise3Citation37 was used to generate Amplicon Sequence Variants (ASVs). Sequences that occurred less than ten times were discarded and sequences with 99% similarity present in negative control samples were removed prior to diversity analyses in order to avoid misleading conclusions.Citation38 The SINTAX algorithmCitation39 with RDP training set v18 reference databaseCitation40,Citation41 was used to annotate taxonomic information. The ASV table was sub-sampled randomly (the smallest number of reads in the sample) to obtain equal sequencing depths among samples. The rarefaction analysis, Observed species, Chao1, ACE, Shannon diversity estimators, Simpson’s diversity index, and Goods’ coverage were measured by R softwareCitation42 with the vegan package.Citation43 Principal coordinates analysis (PCoA), and permutational multivariate ANOVA (PERMANOVA) tests were conducted to evaluate differences in community structures between the samples based on weighted Unifrac distance metrics using the QIIME2 pipeline.Citation36 Linear discriminant analysis effect size (LEfSe)Citation44 with FDR correctionCitation45,Citation46 was used to explore significant differences among groups in the abundance of the taxon. According to the normal distribution and homogeneity of variance, the Analysis of Variance Kruskal-Wallis test was used for statistical analysis, with the significance set at p < 0.05 and q < 0.05 by R package.Citation42 The PICRUSt2 algorithmCitation47 was used to estimate probable Kyoto Encyclopedia of Genes and Genomes (KEGG)Citation48 function of the rat lung microbiome.
Results
Sequencing output and filtering
Experimental rats showed no obvious behavioral or physical changes during the course of the research. The 16S ribosomal RNA genes were sequenced by high-throughput sequencing technology to characterize the microbiota composition and community structure of rat lung microbiota. A total of 3,962,972 microbial sequences (sequences ranged between 40,758 up to 136,338 per animal sample) were detected across samples after discarding the sequences with less than ten.
After filtering for low-quality reads, comparison between the lung samples and negative controls were performed to confirm the effect of potential contamination. As shown in Figure S1, bacterial taxa detected in lung specimens were all collectively distinct from those detected in negative control specimens (p = 0.001 for all comparisons; PERMANOVA). The most abundant ASVs detected in lung specimens and negative controls were very different. These results confirmed that the detection of bacteria in lung specimens was not significantly influenced by potential contamination.
In order to remove only sequences that represented contaminants from the data set, sample sequences that matched any sequence from a negative control at the level of 99% identity over at least 200 bp were removed. In removing single sequences, we also removed ASVs that were present in less than three samples. After the above filtering process, 1,498,257 sequence reads were used for subsequent analyses and resulted in 2677 ASVs (Table S1). Collectively, these sequences represented 21 phyla, 52 classes, 74 orders, 159 families, and 334 genera. The pattern of the most abundant ASVs of the lung samples and the negative controls are similar.
Microbial richness and diversity (alpha diversity)
To evaluate alteration in the microbiota community structure between the groups, the microbial alpha diversity was compared as shown in . Rarefaction curve can verify whether the obtained sequencing data cover all taxa in the sample. As shown in , the rarefaction curve of each group tended to be flat which suggested that the sequencing depth was sufficient to reflect more than 99.3% (Table S1) of the microbiome taxa present. The number of samples extracted was appropriate, and the depth of sequencing data basically included enough taxa of the samples. Rank-abundance curve can represent the abundance and evenness of taxa in the samples. As depicted in , the curve of control group (LC) dropped faster than that in ampicillin groups particularly the LA5 group, indicating lower taxa abundance in LC group.
Figure 1. Alpha diversity comparison between the lung different groups. (A) Rarefaction curve for microbial communities in groups. (B) Rank-abundance distribution curve for microbial communities in groups. (C) The comparison of gut microbiota alpha diversity between each group, including Chao1, ACE, Shannon, and Simpson. Starred samples. Pairwise significance was determined by (C) pairwise and corrected for multiple comparisons using the Benjamini–Hochberg method. Significance key: *p ≤ 0.05; **p ≤ 0.01.
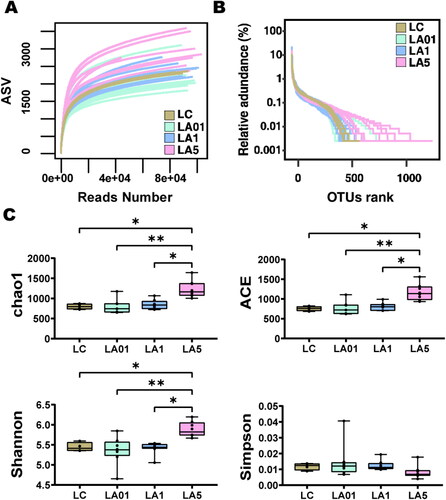
displayed the alpha diversity including Observed species, Chao1, ACE, Shannon, Simpson, and Goods’ coverage (Table S1), of lung microbiota in the four groups of rats administered different concentrations of antibiotics and controls. The result indicated that alpha diversity of the lung microbiota in LA5 group was higher than that of other groups including LC, LA01, and LA1 groups in terms of the richness index, indicating the increasing microbial taxa richness in rats challenged with 5 mg/mL ampicillin (). The Simpson index in LA5 group was lower than in other groups which also represent the higher microbial taxa richness in LA5 group as the above other alpha diversity indexes, though the difference was not significant in statistical analysis. However, when comparing the other ampicillin concentration groups (LA01, LA1) with control group (LC), no significant difference was found in Chao1, ACE, Shannon, and Simpson indexes.
Microbial community composition (beta diversity)
The weighted UniFrac distance metric was created from the subsampled even ASV table to explore structural differences in bacterial communities among the four groups. Principal coordinate analysis (PCoA) showed partial separation of microbiota communities by group (). Samples derived from the LA5 (p = 0.001) exhibited a distinct microbial structure and were completely separated from the LC control group. While microbiota communities in the LC, LA01, and LA1 groups were clustered closely, meaning that their bacterial communities were similar to each other, but separated from those in the LA5 group. The first three principal axes (PC1, PC2, and PC3) explained the most considerable variation of 51.99%. PERMANOVA test confirmed a significant association between the different concentrations of ampicillin treatment groups and bacterial community structures (). The PERMANOVA comparing all modes of ampicillin treatment groups to non-ampicillin group showed a significant difference (p = 0.001), supporting the view that the significant result of the PERMANOVA was due to true differences induced by antibiotic treatment, and could not be explained by differences in dispersion.
Figure 2. Lung microbiome composition in rats of different groups. (A) The microbiome composition of Lung tissue clusters by different concentrations of ampicillin together by principal component analysis of the ASVs using weighted Unifrac distances. The resulting p values and significance keys are tabulated. Individual data points represent specimens grouped by sample or control type. The top 1% relative abundance of detected phyla (B) and genera (C) in all groups. The y-axis denotes relative abundance (%). *p ≤ 0.05; **p ≤ 0.01.
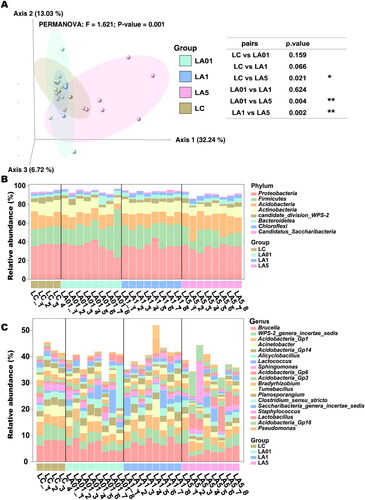
According to 16S rRNA gene sequences, 19 phyla, 41 classes, 65 orders, 127 families, and 216 genera were identified. At the phylum level (), Proteobacteria (35.08%), Firmicutes (20.19%), Acidobacteria (14.91%), Actinobacteria (11.46%), candidate_division_WPS-2 (3.72%), Bacteroidetes (3.51%), Chloroflexi (2.72%) and Candidatus_Saccharibacteria (1.31%) dominating in both the ampicillin group (93.87%), and the untreated group (90.02%). At the genus level (), Brucella (6.02%), WPS-2_genera_incertae_sedis (3.72%), Acidobacteria_Gp1 (3.65%), Acinetobacter (2.46%), Acidobacteria_Gp14 (2.33%), Alicyclobacillus (2.15%), Sphingomonas (1. 80%), Acidobacteria_Gp6 (1.79%), Acidobacteria_Gp3 (1.61%), Bradyrhizobium (1.55%), Tumebacillus (1.51%), Planosporangium (1.47%), Clostridium_sensu_stricto (1.32%), Saccharibacteria_genera_incertae_sedis (1.31%), Staphylococcus (1.24%), Lactobacillus (1.18%), Acidobacteria_Gp16 (1. 16%) and Pseudomonas (1.16%) was dominated (39.11% in the ampicillin group, and 42.22% in the non-ampicillin group).
To further determine which taxa were significantly affected, we performed statistical analysis employing the relative abundance at the phyla to genera levels ( and Citation3). As the LA01 and LA1 groups had no significant difference from the LC group of untreated lungs, indicating that ampicillin needed to reach a certain concentration to cause changes in the microbiota, we compared the LA5 group to find the taxon that potentially be influenced by the ampicillin. LEfSe analyses were conducted to detect the differences between the LC group and ampicillin-treated groups at the ASV, genus, or higher level. Comparison of the LC control and LA5 groups showed the presence of 97 taxa (including 36 genera) with significant differences (). At the genus level, Acidobacteria_Gp16, Ramlibacter, Nitrospira, Gaiella, Aggregatilinea, Latescibacteria_genera_incertae_sedis, and Reyranella were significantly increased in the LA5 group versus the LC control group, whereas Brucella, WPS_2_genera_incertae_sedis, Acinetobacter, Sphingomonas, Acidobacteria_Gp14, Tumebacillus, Acidobacteria_Gp2, Acidobacteria_Gp2, Neobacillus, and Ornithinimicrobium, Virgibacillus were significantly decreased. Besides, there were 8 taxa at the class level, 11 taxa at the order level, and 12 taxa at the family level that were significantly different when compared to the LC with LA5 groups.
Figure 3. Linear discriminant effect size (LEfSe) analyses comparing differentially abundant taxa between different groups. Microbes associated with the effect of different concentrations of ampicillin are shown. (A) Taxonomies differentiating bacterial microbiota in non-ampicillin control treated group (LC) and 5 mg/ml ampicillin treated (LA5) groups, LC and 0.1 mg/ml ampicillin treated (LA01) groups (B), LC and 1 mg/ml ampicillin treated (LA1) groups.
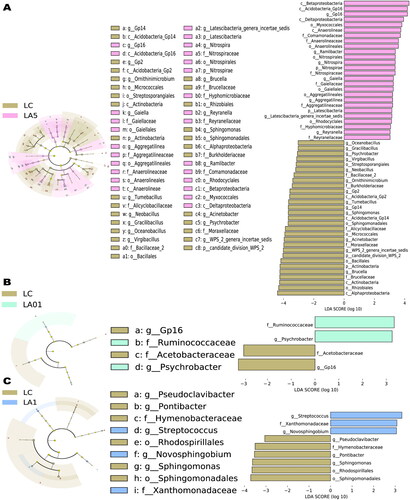
Though the whole microbial community structure, represented by the alpha and beta diversity indexes, had not significantly different between the LA01/LA1 and LC groups, there were still some taxa that were significantly different between them determined by LEfSe analysis. In our comparison of the LA01 and LC groups, the abundances of a total of four taxa (related to two genera) were found to be significantly changed (). At the genus level, Psychrobacter was significantly increased in the LA01 group as compared with the LC group, whereas Actinoallomurus and Acidobacteria_Gp16 were significantly decreased. Finally, in comparison of the LA1 and LC groups, the abundances of a total of nine taxa (including five genera) were found to be significantly changed (). At the genus level, Streptococcus and Novosphingobium significantly increased in the LA1 group as compared with the LC group, whereas Pontibacter, Sphingomonas, and Pseudoclavibacter were significantly decreased.
Differences gene function in all rats lung microbiota samples
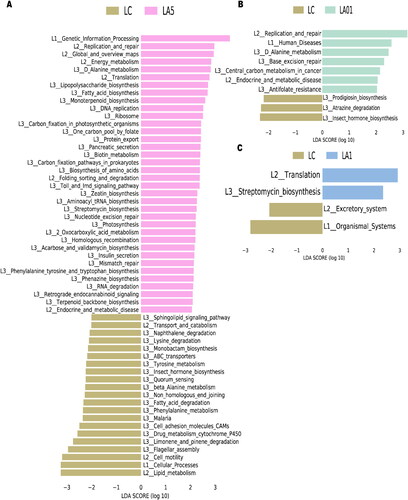
The PICRUSt2 algorithm was further used to analyze the effects of ampicillin on lung microbiota function. A total of 10,543 KEGG orthologs (KOs)Citation48 and 382 pathways were detected. KEGG pathway comparisons were further performed to explore potential differences in the functional composition of the microbiome of antibiotic treatment groups versus no-antibiotic treatment controls by LEfSe analysis. We found that rat lung microbiota samples from the LA5 and LC control groups showed different KEGG profiles. The Drug metabolism cytochrome P450 (map00982), Phenylalanine metabolism (map00360), Fatty acid degradation (map00071), Non_homologous end joining (map03450) and beta-Alanine metabolism (map00410) had significantly lower inferred abundances in the LA5 group than in the LC control group (). The Base excision repair (map03410) and Central carbon metabolism in cancer (map05230) were up-regulated in the LA01 group than in the LC control group (), and the Streptomycin biosynthesis (map00521) had significantly higher inferred abundances in the LA1 group than in the LC group ().
Figure 4. Comparing Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways between different groups with Linear discriminant effect size (LEfSe) analyses. The list of LEfSe provides features that were differential among conditions of biological significance, ranking them according to the effect size. (A) Differential PICRUSt2 predicted KEGG pathways between LC and 5 mg/ml ampicillin treated (LA5) groups; 0.1 mg/ml ampicillin treated (LA01) groups (B); 1 mg/ml ampicillin treated (LA1) groups (C).
Discussion
The present study investigated the effects of ampicillin aerosolization on lung microbiota of Sprague-Dawley rats. Results of this study demonstrated that ampicillin could modify lung community compositions by the long-term low-dose nebulization of antibiotics at a certain concentration of LA5. Brucella, Acinetobacter, Acidobacteria_Gp14, Sphingomonas, Tumebacillus, and Acidobacteria_Gp16 were the dominant and significantly different taxa between the LA5 and LC groups ( and ). Furthermore, alterations were identified in potential metabolites associated with Drug metabolism cytochrome P450 (map00982), Phenylalanine metabolism (map00360), Fatty acid degradation (map00071), Non_homologous end joining (map03450), beta-Alanine metabolism (map00410) and so on ().
The bacterial signal was significantly different between the lung samples and the negative control samples (Figure S1). Potential contamination was eliminated from the data set to facilitate comparison between the different lung sample groups to thus avoid the contamination overwhelming the microbial signal for the low bacterial biomass lung tissue samples.Citation26,Citation49 We used aerosolization to give the ampicillin to rats since it could achieve high antibiotic concentrations in the lower respiratory tract to maximize bacterial killing with minimal systemic side effects and had the minimum effect on gut microbiota,Citation18 avoiding the gut dysbacteriosis that is related to lung microbiota.Citation25,Citation50 Proteobacteria, Firmicutes, Acidobacteria, and Actinobacteria were the dominant phyla in LC group of healthy rats, which was consistent with previous reports.Citation51,Citation52 These results indicated that aerosolization with sterile normal saline (control group in this study) did not change the microbiota composition in comparison to native lungs which was also confirmed by other researchers.Citation51 Moreover, there were no significant differences in lung microbiota between LC, LA01, and LA1 groups, which indicated that only the concentration of ampicillin reached enough concentration that could influence the lung microbial compositions and the lung microbial structure could remain relatively stable for external disturbance.
Antibiotic therapy is often considered to diminish the microbial diversity and abundance, but we found that the alpha diversity index of rat lung microbiome in LA5 group was significantly higher than in other groups, and there was no significant difference in other antibiotic treatment groups versus no-antibiotic treatment controls groups. This phenomenon has been noticed in previous research.Citation53,Citation54 It is likely attributable to the formation of communities of antibiotic-resistant bacteria after the long-term ampicillin treatment. And the differentiated taxon was increased with the increasing antibiotic concentration (). The previous study found after the ampicillin treatment, the diversity of culturable strains decreased at the first 5 days but then the diversity increased with the long-term ampicillin treatment.Citation17 This result was consistent with our study that the long-term ampicillin treatment may gradually enhance the antibiotic-resistant and the microbial diversity increased. The further study should also sample the short-term ampicillin treatment lung tissue to evaluate the microbial diversity. LEfSe revealed that Acidobacteria_Gp16, Ramlibacter, Nitrospira, Gaiella, and so on were the most unique microbes in the LA5 of high concentration ampicillin exposed group (). The above taxon that enriched in the LA5 group mainly belonged to the phyla of Proteobacteria, Acidobacteria, and Actinobacteria. These three phyla have been reported to be the hosts of most antibiotic resistance genes (ARGs) and have the potential resistance to the ampicillin.Citation55–58 We speculated that the shift of the taxon commonly induced by antibiotic treatment, rather than specific taxa, is relevant to the reduced immunosuppression.Citation25,Citation59
Also, the other reason may be that the balance of lung microbiota was disturbed by the certain ampicillin concentration, which increased the risk of bacterial infection and thereby increased microbial diversity. The long-term antibiotics treatment would kill certain bacteria and disturb the lung microbiota balance at the same time. The whole microbial diversity would change with the dysbiosis, including the chance of increasing diversity by the external bacterial infection. However, it is unclear whether antibiotic disturbance induced lung inflammation and then further aggravated the microbiome is altered, or antibiotic disturbance induces microbiome changes and then inflammation occurs. We will explore the relationship between inflammation and microbial changes after antibiotic exposure in future.
In our study, the Proteobacteria, candidate_division_WPS-2, Candidatus_Saccharibacteria, Bacteroidetesand, and Chloroflexi phyla and Bradyrhizobium, Planosporangium, Saccharibacteria_genera_incertae_sedis, Lactobacillus, Acidobacteria_Gp1,Acidobacteria_Gp3, and Alicyclobacillus genera were similar in antibiotic treatment groups versus no-antibiotic treatment controls groups, which was inconsistent with previous studies suggesting that lower microbial diversity favors Proteobacteria abundance,Citation9,Citation60 as the Proteobacteria abundance was not significantly different. Interestingly, we observed no relationship between microbial diversity and Proteobacteria abundance that was consistent with the study of the effects of smoking on the lower respiratory tract microbiome in mice.Citation61 In other words, although the microbial diversity index of LA5 was higher than in other groups (), the relative abundance of Proteobacteria had no significant difference (). And the administration of different concentrations of ampicillin led to significant differences in the relative abundance of above taxon ( and ). However, in the lung of healthy humans, Bacteroidetes and Firmicutes were the two most abundant bacterial phyla, and changes in their abundance in rat lungs can reflect, to some extent, the internal conditions of the human body.Citation62 This distinction is related to the fact that humans and rats are different taxa.
The functional predictions suggested that the administration of different concentrations of ampicillin led to changes in the metabolic pathways of lung microbiota in rats (). Bacterial fitness was closely related to the ability of bacteria to absorb nutrients or amino acids provided by their respective host ecological niches, because they required energy and carbon to grow and replicate.Citation63 Consequently, pathogenic bacterial physiology must be adapted to these different physiological conditions to ensure the expression of adaptive and virulence factors.Citation64 The microbial community changed dynamically depending on the lung condition, followed by pneumococcal infection.Citation65 Compared with the different groups, LA5 showed more dramatic changes in the lung microbiota. showed that D_Alanine_metabolism (map00473), Lipopolysaccharide biosynthesis (map00540), Monoterpenoid biosynthesis (map00902), DNA replication (map03030), Carbon_fixation_in_photosynthetic _organisms (map00710), and so on increased significantly in the LA5 group, some of which were normal microbial metabolic functions (helping digestion and nutrient absorption).Citation53,Citation66 In contrast, the functional pathways involved in Drug metabolism cytochrome P450 (map00982), Phenylalanine metabolism (map00360), Fatty acid degradation (map00071), Non_homologous end joining (map03450), beta-Alanine metabolism (map00410), and so on were reduced; although these alterations in microbiota genetic metabolic pathways were not necessarily associated with lung disease, they deserve the attention of pathology researchers and prescribing clinicians.Citation67
However, the exact interactions between lung microbiota and metabolite changes require further investigation. The KEGG pathway analysis indicated that the dysbiosis of gut bacteria in lung cancer was strongly associated with dysregulation of basic metabolic processes such as energy metabolism and transport.Citation68 In our present study, several metabolism pathways were found to be decreased after ampicillin treatment, such as Fatty acid metabolism, Tyrosine metabolism, Biosynthesis of unsaturated fatty acids, and so on. Therefore, consistent with the previous study,Citation68 we speculated that lung microbiota affected the host via the metabolites or immune system, which provides an improved understanding of perturbations of the microbiome-metabolome interface in lung health and may identify some potential diagnostic and therapeutic targets. However, the function of the microbiome was not necessarily entirely dependent on any of these interactions, and changes in these relationships may affect the lung disease system and cause subtypes.
Although our study yielded some novel insights, there still are some limitations. The gender-specific differences were not investigated which could examine the impact of sex hormones on the rat lung microbiome development. Also, more sampling time points should be considered to track the development of the lung microbiome in rats. At the same time, the effects of fungi and viruses should also be considered.
Conclusions
This study found that a long-term enough concentration of ampicillin exposure by aerosolization can alter the microbial composition in the rat lung. The alpha and beta diversity of Lung microbiota in relatively high concentration ampicillin treated LA5 group were significantly different from and LC control group. The percentage of Acidobacteria_Gp16 in lung tissue changed in the LA5 group after inhalation of ampicillin compared with the LC control group. The D_Alanine_metabolism, Lipopolysaccharide biosynthesis, Monoterpenoid biosynthesis, DNA replication, and Carbon_fixation_in_photosynthetic_organism were elevated and Drug metabolism cytochrome P450, Phenylalanine metabolism, Fatty acid degradation, Non_homologous end joining, and beta-Alanine metabolism were decreased in the LA5 group after atomizing ampicillin compared with the LC control group. Furthermore, defining the composition of the resident microbiome and how microbial communities shift with exposure may help to explain their role in responding to ampicillin. This study could serve as a basis for clinical use of ampicillin in the treatment of respiratory diseases. Subsequent studies are required to determine if these changes in the lung microbiota induced by ampicillin might have an effect on the development and progression of diseases. Also, the impact of the sex hormones, more developmental time points, and fungi and viruses on the lung microbiome of healthy rats, as well as lung microbial functions determining by protein or metabolism levels should be included. At the same time, the effect of the lung microbiome to the gut microbiome and the lung or brain function through the lung-gut or lung-brain microbiota axis should also be considered.
Author contributions
P.R., Y.Z., B.L., and G.L. conceived the study, directed the project, and designed the experiments. H.J. obtained the samples. T.H. performed the microbial sequencing analysis. P.C. interpreted the results, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Consent to participate and for publication
The authors declare no conflict of interest.
Ethics approval
The animal care and experiments in this research were in accordance with the guiding principles for the care and use of laboratory animals recommended by the Chinese Association for Laboratory Animal Science Policy. It was approved by the Institutional Animal Care and Use Committee at the Guangzhou Medical University of China (Guangzhou, China; No. 2019-159).
Supplemental Material
Download MS Word (160.7 KB)Availability of data and material
Sequence data is available at the ENA Accession number: PRJNA862086.
Code availability
Not Applicable.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Thomas S, Izard J, Walsh E, et al. The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res. 2017;77(8):1783–1812. doi:10.1158/0008-5472.Can-16-2929.
- Huang YJ, Nelson CE, Brodie EL, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol. 2011;127(2):372–381.e1-3. doi:10.1016/j.jaci.2010.10.048.
- Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi:10.1371/journal.pone.0008578.
- Borewicz K, Pragman AA, Kim HB, Hertz M, Wendt C, Isaacson RE. Longitudinal analysis of the lung microbiome in lung transplantation. FEMS Microbiol Lett. 2013;339(1):57–65. doi:10.1111/1574-6968.12053.
- Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. doi:10.1146/annurev-physiol-021115-105238.
- Wu BG, Segal LN. The lung microbiome and its role in pneumonia. Clin Chest Med. 2018;39(4):677–689. doi:10.1016/j.ccm.2018.07.003.
- Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014;2(3):238–246. doi:10.1016/s2213-2600(14)70028-1.
- van der Gast CJ, Walker AW, Stressmann FA, et al. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5(5):780–791. doi:10.1038/ismej.2010.175.
- Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the "healthy" smoker and in COPD. PLoS One. 2011;6(2):e16384. doi:10.1371/journal.pone.0016384.
- Poroyko V, Meng F, Meliton A, et al. Alterations of lung microbiota in a mouse model of LPS-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309(1):L76–83. doi:10.1152/ajplung.00061.2014.
- Charlson ES, Diamond JM, Bittinger K, et al. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med. 2012;186(6):536–545. doi:10.1164/rccm.201204-0693OC.
- Blumberg R, Powrie F. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med. 2012;4(137):137rv7. doi:10.1126/scitranslmed.3004184.
- Takesue Y, Yokoyama T, Akagi S, et al. Changes in the intestinal flora after the administration of prophylactic antibiotics to patients undergoing a gastrectomy. Surg Today. 2002;32(7):581–586. doi:10.1007/s005950200105.
- Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect Immun. 2005;73(1):30–38. doi:10.1128/iai.73.1.30-38.2005.
- Russell SL, Gold MJ, Hartmann M, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13(5):440–447. doi:10.1038/embor.2012.32.
- Russell SL, Gold MJ, Willing BP, Thorson L, McNagny KM, Finlay BB. Perinatal antibiotic treatment affects murine microbiota, immune responses and allergic asthma. Gut Microbes. 2013;4(2):158–164. doi:10.4161/gmic.23567.
- Ye M-Q, Chen G-J, Du Z-J. Effects of antibiotics on the bacterial community, metabolic functions and antibiotic resistance genes in Mariculture Sediments during enrichment culturing. JMSE. 2020;8(8):604. doi:10.3390/jmse8080604.
- Zarogoulidis P, Kioumis I, Porpodis K, et al. Clinical experimentation with aerosol antibiotics: current and future methods of administration. Drug Des Devel Ther. 2013;7:1115–1134. doi:10.2147/dddt.S51303.
- Geller DE. Aerosol antibiotics in cystic fibrosis. Respir Care. 2009;54(5):658–670. doi:10.4187/aarc0537.
- Marchisio P, Santagati M, Scillato M, et al. Streptococcus salivarius 24SMB administered by nasal spray for the prevention of acute otitis media in otitis-prone children. Eur J Clin Microbiol Infect Dis. 2015;34(12):2377–2383. doi:10.1007/s10096-015-2491-x.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/s0140-6736(12)61728-0.
- Lopez-Campos JL, Tan W, Soriano JB. Global burden of COPD. Respirology. 2016;21(1):14–23. doi:10.1111/resp.12660.
- Herath SC, Normansell R, Maisey S, Poole P. Prophylactic antibiotic therapy for chronic obstructive pulmonary disease (COPD). Cochrane Database Syst Rev. 2018;10(10):CD009764. doi:10.1002/14651858.CD009764.pub3.
- Facciorusso A, Papagiouvanni I, Cela M, et al. Comparative efficacy of long-term antibiotic treatments in the primary prophylaxis of spontaneous bacterial peritonitis. Liver Int. 2019;39(8):1448–1458. doi: 10.1111/liv.14109.
- Li N, Dai Z, Wang Z, et al. Gut microbiota dysbiosis contributes to the development of chronic obstructive pulmonary disease. Respir Res. 2021;22(1):274. doi:10.1186/s12931-021-01872-z.
- Baker JM, Hinkle KJ, McDonald RA, et al. Whole lung tissue is the preferred sampling method for amplicon-based characterization of murine lung microbiota. Microbiome. 2021;9(1):99. doi:10.1186/s40168-021-01055-4.
- Xu C, Lee SK, Zhang D, Frenette PS. The gut microbiome regulates psychological-stress-induced inflammation. Immunity. 2020;53(2):417–428.e4. doi:10.1016/j.immuni.2020.06.025.
- Ye Z, Wu C, Zhang N, et al. Altered gut microbiome composition in patients with Vogt-Koyanagi-Harada disease. Gut Microbes. 2020;11(3):539–555. doi:10.1080/19490976.2019.1700754.
- Keogh C, Pusceddu M, Kim H-J, Rabasa G, Knotts T, Gareau M. Neonatal dysbiosis has long‐lasting detrimental effects on the microbiota‐gut‐brain axis. FASEB J. 2020;34(S1):1. doi:10.1096/fasebj.2020.34.s1.04489.
- Tang X, Wang W, Hong G, et al. Gut microbiota-mediated lysophosphatidylcholine generation promotes colitis in intestinal epithelium-specific Fut2 deficiency. J Biomed Sci. 2021;28(1):20. doi:10.1186/s12929-021-00711-z.
- Wang P, Zhang L, Liao Y, et al. Effect of intratracheal instillation of ZnO nanoparticles on acute lung inflammation induced by lipopolysaccharides in mice. Toxicol Sci. 2020;173(2):373–386. doi:10.1093/toxsci/kfz234.
- Toyama H, Ichise M, Liow J-S, et al. Evaluation of anesthesia effects on [18F]FDG uptake in mouse brain and heart using small animal PET. Nucl Med Biol. 2004;31(2):251–256. doi:10.1016/S0969-8051(03)00124-0.
- Gargiulo S, Greco A, Gramanzini M, et al. Mice anesthesia, analgesia, and care, Part I: Anesthetic considerations in preclinical research. ILAR J. 2012;53(1):E55–E69. doi:10.1093/ilar.53.1.55.
- de Sena Brandine G, Smith AD. Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res. 2019;8:1874. doi:10.12688/f1000research.21142.2.
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi:10.1093/bioinformatics/btr507.
- Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9.
- Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016;21:081257. doi:10.1101/081257.
- Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi:10.1186/s12915-014-0087-z.
- Edgar RC. SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. 2016;20:074161. doi:10.1101/074161.
- Edgar RC. Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ. 2018;6:e5030. doi:10.7717/peerj.5030.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi:10.1093/bioinformatics/btq461.
- Team RC. R: A language and environment for statistical computing. Updated 2022/06/23. https://www.R-project.org/. Accessed July, 2022.
- Oksanen J, Blanchet FG, Friendly M, et al. Vegan: community ecology package. Updated 2022/04/17. https://CRAN.R-project.org/package=vegan. Accessed July, 2022.
- Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60.
- Nearing JT, Douglas GM, Hayes MG, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13(1):342. doi:10.1038/s41467-022-28034-z.
- Rooks MG, Veiga P, Wardwell-Scott LH, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8(7):1403–1417. doi:10.1038/ismej.2014.3.
- Douglas GM, Maffei VJ, Zaneveld JR, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–688. doi:10.1038/s41587-020-0548-6.
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27.
- Scheiermann J, Klinman DM. Three distinct pneumotypes characterize the microbiome of the lung in BALB/cJ mice. PLoS One. 2017;12(7):e0180561. doi:10.1371/journal.pone.0180561.
- Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. 2013;14(7):646–653. doi:10.1038/ni.2604.
- Finn SMB, Scheuermann U, Holzknecht ZE, et al. Effect of gastric fluid aspiration on the lung microbiota of laboratory rats. Exp Lung Res. 2018;44(4–5):201–210. doi:10.1080/01902148.2018.1482976.
- Li N, He F, Liao B, Zhou Y, Li B, Ran P. Exposure to ambient particulate matter alters the microbial composition and induces immune changes in rat lung. Respir Res. 2017;18(1):143. doi:10.1186/s12931-017-0626-6.
- Cheng X, He F, Si M, Sun P, Chen Q. Effects of antibiotic use on saliva antibody content and oral microbiota in sprague dawley rats. Front Cell Infect Microbiol. 2022;12:721691. doi:10.3389/fcimb.2022.721691.
- Dudek-Wicher RK, Junka A, Bartoszewicz M. The influence of antibiotics and dietary components on gut microbiota. PRZ Gastroenterol. 2018;13(2):85–92. doi:10.5114/pg.2018.76005.
- Jiang X, Ellabaan MMH, Charusanti P, et al. Dissemination of antibiotic resistance genes from antibiotic producers to pathogens. Nat Commun. 2017;8:15784. doi:10.1038/ncomms15784.
- Zhao R, Feng J, Liu J, Fu W, Li X, Li B. Deciphering of microbial community and antibiotic resistance genes in activated sludge reactors under high selective pressure of different antibiotics. Water Res. 2019;151:388–402. doi:10.1016/j.watres.2018.12.034.
- Gao FZ, He LY, He LX, et al. Untreated swine wastes changed antibiotic resistance and microbial community in the soils and impacted abundances of antibiotic resistance genes in the vegetables. Sci Total Environ. 2020;741:140482. doi:10.1016/j.scitotenv.2020.140482.
- Wang Y, Lu S, Liu X, et al. Profiles of antibiotic resistance genes in an inland salt-lake Ebinur Lake, Xinjiang, China: The relationship with antibiotics, environmental factors, and microbial communities. Ecotoxicol Environ Saf. 2021;221:112427. doi:10.1016/j.ecoenv.2021.112427.
- Qiao L, Liu X, Zhang S, et al. Distribution of the microbial community and antibiotic resistance genes in farmland surrounding gold tailings: A metagenomics approach. Sci Total Environ. 2021;779:146502. doi:10.1016/j.scitotenv.2021.146502.
- Huang YJ, Kim E, Cox MJ, et al. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS. 2010;14(1):9–59. doi:10.1089/omi.2009.0100.
- Zhang R, Chen L, Cao L, et al. Effects of smoking on the lower respiratory tract microbiome in mice. Respir Res. 2018;19(1):253. doi:10.1186/s12931-018-0959-9.
- Marsland BJ, Gollwitzer ES. Host-microorganism interactions in lung diseases. Nat Rev Immunol. 2014;14(12):827–835. doi:10.1038/nri3769.
- Dong H, Tan R, Chen Z, et al. The effects of immunosuppression on the lung microbiome and metabolites in rats. Front Microbiol. 2022;13:817159. doi:10.3389/fmicb.2022.817159.
- Härtel T, Eylert E, Schulz C, et al. Characterization of central carbon metabolism of Streptococcus pneumoniae by isotopologue profiling. J Biol Chem. 2012;287(6):4260–4274. doi:10.1074/jbc.M111.304311.
- Chen YW, Li SW, Lin CD, et al. Fine particulate matter exposure alters pulmonary microbiota composition and aggravates pneumococcus-induced lung pathogenesis. Front Cell Dev Biol. 2020;8:570484. doi:10.3389/fcell.2020.570484.
- Deo PN, Deshmukh R. Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol. 2019;23(1):122–128. doi:10.4103/jomfp.JOMFP_304_18.
- Xiao J, Fiscella KA, Gill SR. Oral microbiome: possible harbinger for children’s health. Int J Oral Sci. 2020;12(1):12. doi:10.1038/s41368-020-0082-x.
- Qu Z, Zhang L, Hou R, et al. Exposure to a mixture of cigarette smoke carcinogens disturbs gut microbiota and influences metabolic homeostasis in A/J mice. Chem Biol Interact. 2021;344:109496. doi:10.1016/j.cbi.2021.109496.