
Abstract
Recent research has revealed that airway epithelial calcium-activated chloride channel-1 (CLCA1) is implicated in the inflammation of multiple human respiratory diseases, but the specific role in acute respiratory distress syndrome (ARDS) remains unknown. To investigate the role of CLCA1 in ARDS, 80 participants, including 26 ARDS patients, 26 patients with community-acquired pneumonia (CAP) and 28 control subjects, were enrolled in this study. As the result shows, the level of CLCA1 was significantly increased in ARDS patients and positively correlated with neutrophil infiltration and the poor prognosis of ARDS. Then, the level of CLCA1 also elevated in the LPS-induced ARDS mouse model, and the administration of CLCA1 significantly regulated the phenotypes of ARDS in mice, such as lung injury score, BALF protein concentration, neutrophils infiltration and the secretions of inflammatory factors. Furthermore, administration of CLCA1 substantially altered the phosphorylation of p38 in the ARDS mouse model, whereas repressing the expression of CLCA1 or inhibiting the activation of p38 both alleviated the inflammatory response of ARDS. In summary, CLCA1 was notably correlated with ARDS and exacerbated the ARDS phenotypes through the p38 MAPK pathway.
Introduction
Acute respiratory distress syndrome (ARDS) is an acute lung injury which is characterized by decreased lung compliance, moderate to severe oxygenation impairment, protein-rich pulmonary edema and bilateral pulmonary infiltrates on chest radiographs.Citation1 Pneumonia, mechanical aspiration, and the more recent COVID-19 pandemic are leading risk factors for the aggravated morbidity of pulmonary ARDS.Citation2,Citation3 Over 3 million people are affected by ARDS, and the mortality is approximately 40%.Citation4 Moreover, the prevalence of COVID-19 elevated the ICU mortality to 61.5%, which would increase to 65.7%∼94% if patients received mechanical ventilation.Citation5
Uncontrolled hyperinflammation, also known as cytokine storm, is recognized as a central role in the pathogenesis of ARDS, which is characterized by overwhelmingly systemic inflammation, hyperferritinemia, hemodynamic instability and multi-organ failure.Citation6 This uncontrolled immune response starts from a sudden acute increase in circulating levels of different pro-inflammatory cytokines, including IL-6, IL-1β, TNF- α, and interferon, and orchestrates the influx of various immune cells such as macrophages, neutrophils, and lymphocytes from the circulation into the site of infection.Citation7 The cytokine storm leads to ARDS aggravation and widespread tissue damage, eventually resulting in multi-organ failure and death.Citation8,Citation9 However, the triggering factors and mechanism of cytokine storm are still unclear, whereas the conventional drugs, such as glucocorticoid, nitric oxide and Sivelestat, failed to reduce the mortality of ARDS or to shorten the duration of mechanical ventilation.Citation10–12 Therefore, revealing the mechanism of ARDS and identifying the triggering variables and participating cells would provide theoretical support for the exploration of new drugs and clinical treatments.
Airway epithelial cells, an essential subset of pulmonary structural cells, not only serve as a structure barrier against allergens, pathogens and other environmental factors but are also involved in the immune response by secreting multiple cytokines and inflammatory mediators. Recent researches believed that airway epithelial cells play a triggering and exacerbating role in the inflammatory process of lung disease.Citation13,Citation14 Additionally, a recent study revealed a link that the crosstalk between epithelial and immune cells was associated with the severity of COVID-19 by using single-cell sequencing.Citation15 However, due to the heterogeneity of cells and the complex interaction with other cell subsets, the potential functions of airway epithelial cells in inflammation-mediated ARDS are still unclear. Our study emphasized the airway epithelial and revealed the clued of the ARDS mechanism.
CLCA1 is the first human member of the family of Ca2+-activated Cl-channel proteins and is selectively expressed in goblet cells, mucosal epithelia and other mucin-producing cells in the mucus layer of airways. It is determined that the soluble heterodimer form contains two protease cleavage sites and can be degraded into a 75 kDa amino-terminal protein and a 35 kDa carboxy-terminal protein in the Golgi apparatus.Citation16 Murine CLCA1, also known as Gob-5 or mCLCA3, was recently named in accordance with the human CLCA nomenclature.Citation17 It is believed that CLCA1 acted as a pivotal mediator for several respiratory diseases, such as asthma, COPD, and cystic fibrosis.Citation18–20 Meanwhile, intratracheal instillation of N-CLCA1 to the murine asthma model promoted mucin secretion, airway hyperresponsiveness, and inflammatory cell penetration into the lungs.Citation21 Additionally, the less neutrophilic infiltration and lower concentrations of BALF IL-17 and CXCL-8 were demonstrated in the CLCA knock-out pneumonia mouse model.Citation22 However, the functions of CLCA1 in the pulmonary ARDS are still unclear, and we aimed to discover the role of key ion channel protein CLCA1 in ARDS pathogenesis.
Materials and methods
Participates recruitment
All the participants were adults, and they were recruited in Xijing hospital of Fourth Military Medical University from January 2017 to June 2018, including clinical outpatients and hospitalized patients. The patients’ BALF was obtained from clinical diagnosis and treatment. 26 pulmonary ARDS patients, 26 community-acquired pneumonia (CAP) patients and 28 control subjects with isolated pulmonary nodules or mass lesions on CT imaging were enrolled in this study. According to the criteria of the Berlin Guidelines, ARDS patients were administered at ICU within 48 h of onset, diagnosed as pulmonary infection and received bedside tracheoscopy. According to the diagnosis and treatment of CAP in Chinese adults (2016), CAP patients were diagnosed and received tracheoscopy within 48 h of onset. The control subjects were diagnosed as isolated pulmonary noninfectious nodules on CT imaging, bronchoscope and BALF analyses were prescribed for further identification. Patients with concomitant diseases of COPD, asthma, bronchiectasis, hematological diseases and other chronic diseases were excluded from this study. Patients with long-term smoking history or using systemic corticosteroids in 7 days were also disqualified. This study was approved by the ethics committee of Xijing Hospital and the informed consents were obtained from all patients or their guardians.
Human BALF collection and neutrophils counting
The BALF samples of ARDS and CAP groups were collected at sites of radiographic exudative lesions, whereas the samples of control group were obtained from sites of normal lobes. BALF samples in CAP group and normal controls were collected at baseline, and for ARDS patients, samples were collected in the same lobe at baseline and 7 days after treatment. 5 doses of sterile saline (37 °C) were injected into the bronchoscope working channel, and the collecting rate of lavage fluid was 40%-60%. Lavage solution was filtered by double-layer gauze and then centrifuged at 1200 rpm for 10 min. The supernatant of the centrifuged lavage was collected and stored at −80 °C for the inflammatory cytokines analyses. The BALF cells were resuspended at a density of 5 × 109/L. 100 µL cell suspension was added to the slide and fixed in anhydrous ethanol for 30 min of Reggie’s staining. 200 cells were randomly counted under microscope and the ratio of neutrophils in total cells was calculated.
ARDS mice model and BALF collection
Male C57BL/6 mice (6 ∼ 8 wk) were obtained from the Experimental Animal Center of Air Force Military Medical University and maintained under standard conditions with free access to water and food. The mice were randomly separated into 4 groups and placed in the anesthesia chamber with 5% isoflurane in the air for 5 min before the LPS treatment. And then, the anesthetized mice were exposed to LPS (Sigma Aldrich, St. Louis, MO, USA) intratracheally (0, 50, 100, 200 μg in 50 μL PBS) to construct the ARDS model, the treated mice were grouped and named by drug dose of 2.5 mg/kg, 5 mg/kg, 10 mg/kg LPS. The mice were treated by LPS to imitate the inflammatory phenotype of ARDS, the murine lung sections, the levels of inflammatory cytokines, inflammatory cells infiltration and wet/dry weight ratio were utilized to evaluate the inflammatory level of mice model. Besides, to elucidate the role of CLCA1 in ARDS, mice were treated with 50 μL N-CLCA1 at a concentration of 100 ng/mL (Sino Biological Inc., Beijing, China) or 9 μL CLCA1 blocking antibody (abCLCA1, Abcam, Cambridge, UK) in 5 mg/kg LPS stimulated mice. Murine BALF samples were retrieved 24 h after these treatments, and BALF protein concentration was assessed by BCA kit (Sigma Andrich).
Lung wet weight to dry weigh ratio
Mice of each group were sacrificed at 24 h after LPS treatment. wet weight was recorded immediately, whereas dry weight was recorded after 72h incubation at 60 °C.
Histological staining and lung injury score measuring
The murine lung tissues were fixed in paraformaldehyde solution (4%) at room temperature for 24 h. The lung sections were stained by HE. Semi-quantitative scores of lung injury were calculated according to the following index: alveolar septum thickening, hemorrhage, alveolar hyperemia, and neutrophil infiltration. Each index was graded from 0 to 5 points by two independent investigators, and the total scores of the four indices were recorded as lung injury scores.
CLCA1 immunohistochemical staining
Paraffin-embedded specimens were deparaffinized in xylene, rehydrated through a decreasing ethanol gradient, and rinsed in PBS. The immunohistochemistry staining for CLCA1 (1:100, Invitrogen, CA, USA) was performed according to the procedures of the immunohistochemical staining kit (Yeasen, Shanghai, China).
HBE cell culture and treatment
Human bronchial epithelial cell line 16HBE was grown in DMEM medium with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in a cell incubator with 5% CO2. Cells were plated in 48-well plates at a density of 2 × 105 cells/well, and 200 nM siRNA/NC combined with Lipofectamine 3000 (Thermo Fisher, MA, USA) were transfected into cells. CLCA1 siRNA: 5′-AAG UUA GUG AGG UAG GCU GGG AAC C-3′; negative control: 5′-GGU UCC CAG CCU ACC UCA CUA ACU U-3′. After 24h of incubation, the transfected cells were incubated with 5 μg/mL LPS for 24h. Meanwhile, the other groups of cells were treated with recombinant N-CLCA1 (10 ng/mL) or p38 MAPK inhibitor SB203580 (50 nM) plus LPS (5 μg/mL) for 24h as well. The expression of CLCA1 was evaluated using western blot.
Enzyme linked immunosorbent assay (ELISA)
According to the manufacturer’s protocol, the collected human and murine BALF samples were centrifuged at 800 g for 10 min, and the supernatants were collected for ELISA analysis. CLCA1 in human BALF, as well as CXCL1, CXCL2, TNF-α, and IL-1β in 16HBE cell supernatant, were measured using ELISA kits (Shanghai Enzyme-linked Biotechnology Co Ltd, Shanghai, China). Murine CXCL1, CXCL2, TNF-α, and IL-1β were detected using ELISA kits purchased from R&D System (Minneapolis, MN, USA).
Western blot
The protein samples were extracted from mouse lung tissues and treated cells by lysis buffer, and the concentration was measured by BCA kit. Samples with equal amounts of protein were loaded into 10% SDS polyacrylamide gels and transferred to PVDF membranes (Millipore, MA, USA). The membranes were blocked with 1% BSA in TBST for 1h at room temperature and then incubated overnight at 4 °C with 1:1000 dilutions of p38, p-p38 and GAPDH primary antibodies (Abcam, Cambridge, UK) separately, followed by the incubation with 1:3000 dilutions of HRP-labeled secondary antibody for 30 min. After washing in TBST three times, the membranes were exposed to ECL solution (Bio-Rad, CA, USA) and imaged by chemiluminescence.
Statistical analysis
Statistical analysis was performed using SPSS 22.0 (IBM Software, NY, USA). All continuous data were presented as means ± SD. One-way ANOVA with LSD post hoc test was performed to compare the multiple groups. P < 0.05 is considered significant.
Results
Increased CLCA1 is associated with the poor prognosis of pulmonary ARDS
To investigate the role of CLCA1 in ARDS, we enrolled the patients with ARDS or CAP as well as normal controls. The patients with ARDS had considerably greater levels of CLCA1 (5.03 ± 1.55 ng/L vs 50.01 ± 20.72 ng/L, P < 0.05), while the CAP participants also showed significantly higher levels of CLCA1(22.35 ± 11.85 ng/L, P < 0.05) (). The central role of neutrophil in ARDS pathology was verified by the BALF cell counting, and the result showed that the ratio of neutrophils in ARDS group was higher than CAP and control group (). Furthermore, there was a significant correlation between the ratio of neutrophils and the level of CLCA1 in patients’ BALF (r = 0.724, p < 0.01) (), indicating CLCA1 was implicated with the pathogenesis of ARDS. Additionally, the results revealed that CLCA1 was expressly correlated with the clinical mortality of ARDS (). According to the clinical outcomes, ARDS patients were classified as survivors (n = 14) and non-survivors (n = 12). The age, gender composition and evaluating indices of disease severity, such as PaO2/FiO2 ratio, APACHE and SOFA scores, both were listed in . More importantly, significantly higher levels of CLCA1 were shown in non-survivors (38.12 ± 16.27 ng/L vs 52.29 ± 17.46 ng/L, P < 0.05). Meanwhile, at 7 days after the onset, the levels of CLCA1 were decreased in survivors, whereas it was elevated in non-survivors at the same time. All the results elucidated the link between the level of CLCA1 and the poor prognosis of ARDS.
Figure 1. The level of CLCA1 is increased in ARDS patients.
80 participates, including 26 patients with ARDS, 26 patients with CAP and 28 control subjects with isolated pulmonary nodules or mass lesions, were enrolled in this research. A. The level of CLCA1 in human Bronchoalveolar lavage fluid (BALF) detected by ELISA, *P < 0.05, compared with control; #P < 0.05, compared with CAP. B. The ratio of neutrophils in human Bronchoalveolar lavage fluid (BALF) calculated by BALF cells counting, *P < 0.05, compared with control; #P < 0.05, compared with CAP. C. Liner regression analysis for the ratio of neutrophils and the level of CLCA1. D. The level of CLCA1 in survivors and non-survivors of ARDS detected by ELISA, *P < 0.05, compared with survivors; #P < 0.05, compared with the baseline of survivors.
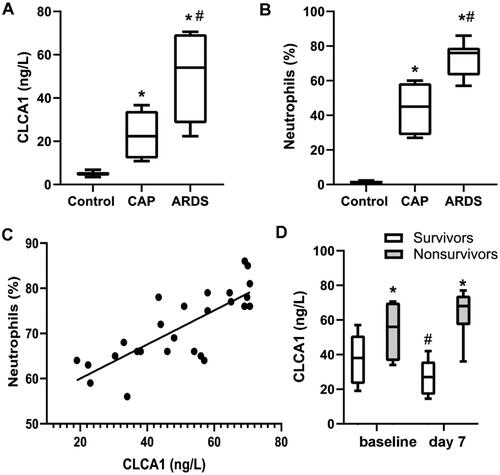
Table 1. Clinical characteristics of the ARDS patients.
The level of CLCA1 is elevated in ARDS mouse model
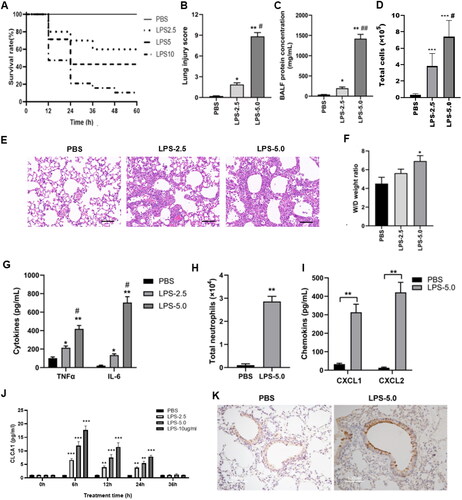
Since the level of CLCA1 was evidently correlated with the progression of ARDS in patients’ samples, the level and function of CLCA1 were further clarified in ARDS mouse models. Three different LPS doses were utilized to construct the mouse ARDS model. Over 90% of mice that received 10 mg/kg LPS was dead at 60h after stimulation (). The results indicated that LPS increased the mortality of mice in a dose-dependent manner, and the 2.5 mg/kg and 5.0 mg/kg doses would be utilized in subsequent studies. The doses of LPS at 2.5 mg/kg and 5.0 mg/kg both could effectively exacerbate the lung injury and significantly elevate the indices of inflammation in mice, including lung injury score (), BALF protein () and the inflammatory cytokines TNF-α and IL-6 (). Furthermore, the 5.0 mg/kg group exhibited significantly increased inflammation and thickened alveolar septum than the 2.5 mg/kg group (). But only the dose of LPS at 5.0 mg/kg could significantly aggravate the W/D weight ratio of mouse model (). Additionally, LPS stimulation significantly enhanced neutrophils recruitment and chemokine release in mice (-21). Moreover, LPS stimulation dramatically boosted the level of CLCA1 in mice at 6h after stimulation. Subsequently, the level of CLCA1 gradually decreased until 36h, when it was marginally higher than the control (). The positive immunohistochemical staining indicated the notable expression of CLCA1 in ARDS mouse model ().
Figure 2. LPS stimulation elevates the level of CLCA1 in ARDS mouse model.
A. The survival rate of LPS-stimulated mice, mice were intratracheally administrated with different concentrations of LPS (0, 2.5, 5.0 and 10.0 mg/kg), n = 12. B. The lung injury score of LPS-stimulated mice, n = 6, *P < 0.05, **P < 0.01, compared with PBS group, #P < 0.05, compared with LPS-2.5 group. C. The BALF protein concentration of LPS-stimulated mice, n = 6, *P < 0.05, **P < 0.01, compared with PBS group, ##P < 0.01, compared with LPS-2.5 group. D. The total BALF cells of LPS-stimulated mice, n = 6. ***P < 0.001, compared with PBS group, #P < 0.05, compared with LPS-2.5 group. E. H&E staining of murine lung sections, scale bar 100μm. F. The lung W/D weight ratio of LPS-stimulated mice, n = 6, *P < 0.05, compared with PBS group. G. the level of TNF-α and IL-6 in LPS stimulated mice detected by ELISA, *P < 0.05, **P < 0.01, compared with PBS group; ## P < 0.01, compared with LPS-2.5 group. H. The number of BALF neutrophils in LPS stimulated mice, n = 6, **P < 0.01. I. The level of CXCL1 and CXCL2 in LPS stimulated mice detected by ELISA, n = 6, **P < 0.01. J. The level of CLCA1 in LPS stimulated mice detected by ELISA, n = 6, **P < 0.01, compared with PBS group. K. The murine lung sections were dyed by immunohistochemistry stain, the brown area is specific staining of CLCA1, scale bar 100 μm.
CLCA1 is pivotal for the inflammatory response and lung injury in ARDS mouse model
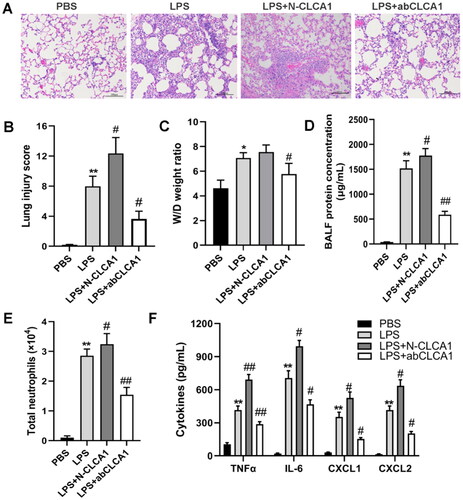
To investigate the role of CLCA1 in the pathogenesis of ARDS, the synthetic N-terminal CLCA1 and CLCA1 blocking antibody were intratracheally administered to ARDS mice, and the mice were sacrificed at 24h after administration. The H&E-stained sections indicated that N-CLCA1 exacerbated inflammatory cells infiltration and alveolar septum thickness, whereas CLCA1 blocking antibody clearly reduced the pulmonary abnormalities of ARDS (). Similarly, the evaluating indices of ARDS severity, including lung injury score, W/D weight ratio and BALF protein, both were modulated by N-CLCA1 and CLCA1 blocking antibody (). Meanwhile, the inflammatory cells and factors, such as neutrophils, IL-6, TNF-α, CXCL1 and CXCL2, were substantially induced by N-CLCA1 and demonstrably decreased by CLCA1 blocking antibody (). All the results indicated that CLCA1 is essential for the LPS-induced ARDS phenotypes in mice.
Figure 3. CLCA1 Exacerbates pulmonary inflammation, lung injury and inflammatory cytokines release in ARDS mouse model.
A. H&E staining of murine lung sections, scale bar 100μm. B. The lung injury score of ARDS mouse model, n = 6, *P < 0.05, compared with PBS group; #P < 0.05 compared with LPS group. C. The lung wet/dry weight ratio of ARDS mouse model, n = 6, *P < 0.05, compared with PBS group; #P < 0.05 compared with LPS group. D. The BALF protein concentration of ARDS mouse model, n = 6, **P < 0.01, compared with PBS group; #P < 0.05, ##P < 0.01, compared with LPS group. E. The number of BALF neutrophils in ARDS mouse model, n = 6, **P < 0.01, compared with PBS group; #P < 0.05, ##p < 0.01, compared with LPS group. F. The level of inflammatory cytokines in ARDS mouse model detected by ELISA, including IL-6, TNF-α, CXCL1 and CXCL2, n = 6, ** p < 0.01, compared with PBS group; #p < 0.05, ##p < 0.01, compared with LPS group.
CLCA1 exacerbates the inflammatory response via p38 MAPK pathway
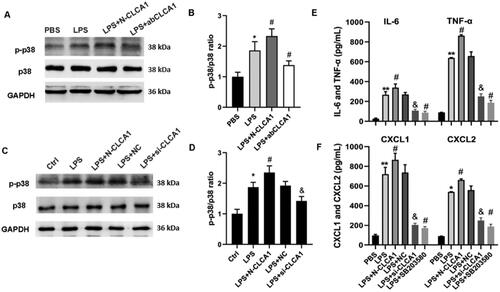
Since the p38 MAPK pathway is essential in the progression and pathogenesis of ARDS,Citation23 the role of p38 in CLCA1 modulated inflammatory response was verified in mice and human bronchial epithelial cells. The expression of phosphorylated and total p38 in ARDS mouse model was evaluated by western blot, and the ratio of p-p38/p38 was significantly increased in ARDS mouse model. Similarly, the ratio of p-p38/p38 was substantially elevated by N-CLCA1 and notably decreased by CLCA1 antibody (). To verify the regulating role of p38 in CLCA1 exacerbated ARDS phenotypes, the human bronchial epithelial cells 16HBE were employed in subsequent studies. Corresponding to the results of ARDS mouse model, the ratio of p-p38/p38 was clearly elevated by N-CLCA1 and decreased by siRNA of CLCA1 (). Furthermore, the inflammatory factors in 16 HBE cells, such as IL-6, TNF-α, CXCL1 and CXCL2, were detected by ELISA. The results confirmed the promoting role of CLCA1 in the secretion of inflammatory factors of ARDS. Conversely, repressing the expression of CLCA1 or inhibiting the activation of p38 both notably alleviated the inflammatory response (). The results indicated that CLCA1 exacerbates the inflammatory response of ARDS via p38 MAPK pathway.
Figure 4. CLCA1 Modulates the release of inflammatory factors via p38 MAPK activation.
A. P38 expression was detected by western blot in ARDS mouse model. B. The ratio of p38 expression western blot in ARDS mouse model, n = 6, **P < 0.01, compared with PBS group; #p < 0.05, compared with LPS group. C. P38 expression was detected by western blot in 16HBE cells. D. The ratio of p38 expression western blot in ARDS mouse model, n = 6, *P < 0.05, compared with PBS group; #P < 0.05, compared with LPS group; &P < 0.05, compared with LPS + NC group. E. The concentrations of TNF-ɑ and IL-6 in 16HBE cells were detected by ELISA, n = 6, **P < 0.01, compared with PBS group; #P < 0.05, compared with LPS group; &P < 0.05, compared with LPS + NC group. F. The concentrations of CXCL1 and CXCL2 16HBE cells were detected by ELISA, n = 6, *p < 0.05, **P < 0.01, compared with PBS group; #P < 0.05, compared with LPS group; &P < 0.05, compared with LPS + NC group.
Discussion
This study demonstrated that CLCA1 notably provoked pulmonary inflammatory responses and associated with lung injury in ARDS, thereby identified a promising therapeutic target of ARDS. CLCA1 is highly expressed in human and murine mucosal epithelial cells and involved in chloride current and mucin expressionCitation20 and multiple studies have highlighted the role of CLCA1 in chronic respiratory diseases. Clinical research has indicated the differentially methylation of CLCA1 was crucially implicated with childhood lung function impairment, asthma, and COPD across the life course.Citation24 Furthermore, CLCA1 was highly correlated with IL-9 expression in the bronchial biopsies of asthmatic patients,Citation25 and the overexpressed CLCA1 was closely associated with airway hyperresponsiveness and infiltration of inflammatory cells in asthmatic mice.Citation21 Recent results of mRNA microarray revealed that mRNA of CLCA1 was the most upregulated gene transcript (>100-fold) in the diisocyanate-exposed asthmatic mouse model.Citation26 Our previous findings indicated that the DNA vaccine-induced CLCA1 antibodies remarkably reduce the number of eosinophils and mast cells in the BALF of asthmatic mice.Citation27 According to the above evidences, we assumed the level of CLCA1 is closely related to the inflammation in chronic lung disease.
However, few researches focused on the role of CLCA1 in the pathogenesis of acute lung injury or ARDS. Dietert K et al. reported that reduced neutrophilic infiltration and decreased level of IL-17 and CXCL-8 were detected in the CLCA1 knockout pneumonia mice induced by Staphylococcus aureus.Citation22 Additionally, LPS stimulation promoted the expression of CLCA1 in human airway mucoepidermoid cells.Citation28 Meanwhile, the synthesized CLCA1 induced the production of IL-1β, IL-6, TNF-α and IL-8 from airway macrophages.Citation29 These researches suggested the close relationship between CLCA and acute lung inflammation. In our research, the result clearly indicated CLCA1 was positively correlated with the neutrophil infiltration in pulmonary ARDS, which provide a proof to demonstrate that CLCA1 was participated in the development of ARDS. Furthermore, the administration of CLCA1 significantly regulated the LPS-stimulated phenotypes of mice. But the detailed etiological factors and the mechanisms of CLCA1 production in ARDS or CAP are still under further investigation.
The airway epithelium is the first barrier of the respiratory system to against the environmental factors by secreting airway mucus and pro-inflammatory factors and recruiting inflammatory cells. This study uncovered a promising target in airway epithelium to impede the cytokine storm in ARDS. Additional N-CLCA1 treatment amplified the production of inflammatory factors induced by LPS, including IL-6, TNF-α, CXCL1 and CXCL2. But most importantly, CXCL1 and CXCL2 are key chemokines to promote the motivation and migration of neutrophils, which demonstrated the regulating role of CLCA1 in the infiltration of immune cells. A recent study revealed the link that the crosstalk between epithelial and immune cells was associated with the severity of COVID-19 by using single cell sequencing.Citation15 Centeio et al. also found that CLCA1 was elevated in IL-13-treated human airway epithelial cells.Citation21,Citation30 CLCA1 activates airway macrophages to secret multiple inflammatory cytokines including IL-1β, IL-6, TNF-α and IL-8.Citation29 Therefore, we supposed that the elevated CLCA1 is an excessive response of the airway epithelium to initiate or exacerbate inflammation of ARDS, and CLCA1 knockout mice need to be adopted to verify this hypothesis in further research.
p38 MAPK plays a pivotal role in the release of inflammatory mediators and the infiltration of inflammatory cells, contributing to the acute lung injury in ARDS. Previous studies revealed that JNK and p38 MAPK participated in sepsis-induced murine lung injury, and their inhibitors improved lung permeability, attenuated system inflammation, and further alleviated lung injury.Citation31 Besides, p38 MAPK is also involved in the burn wound or pancreatitis induced acute lung injury and pneumonia,Citation32,Citation33 and it was recognized that the p38 MAPK inhibitor has great potential for clinical treatment.Citation34 Alevy et al. reported that CLCA1 activated MAPK13 cascade to promote airway mucus production in human airway epithelial and COPD mice.Citation20,Citation35 This study demonstrated that p-p38 expression was significantly elevated in N-CLCA1 treated in ARDS mouse model and airway epithelial cells, which indicated that p38 is the pivotal mediator in CLCA1 promoted lung injury and inflammatory response of ARDS. Nevertheless, this work was limited to examining the levels of p38 in mouse lung tissue, additional research is required to completely investigate the precise mechanism of the p38 MAPK pathway and the regulating role of p38 in CLCA1-mediated inflammations of ARDS.
In conclusion, our study demonstrated that CLCA1 was significantly upregulated in ARDS patients and associated with neutrophil infiltration and poor prognosis of ARDS. Additional N-CLCA1 aggravated murine lung injury and pulmonary protein deposition, as well as the production of inflammatory cytokines. Besides, N-CLCA1 initiates inflammatory response in human bronchial epithelial cells via the p38 MAPK signaling pathway. However, the mechanism and mediators were mainly assessed in lung tissue from LPS-induced ARDS mice model, given the limited results from cells model and patients’ samples, more research is still required to completely comprehend the detailed mechanism of CLCA1 activated p38 MAPK pathway.
Ethical approval
The study was reviewed and approved by the ethics committee of Xijing Hospital of Fourth Military Medical University (XJYYLL-2015111).
Authors’ contributions
Conceptualization, LS; Methodology, LZ, TZ, YC, JL, ZC, and XG; Writing-Original Draft, XL; Writing-Review & Editing, XL, WW, and LS; Funding acquisition, XL and LS. All authors contributed to the article and approved the submitted version.
Disclosure statement
No potential conflict of interest was reported by the author(s). The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Additional information
Funding
References
- Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration. 2017;93(3):212–225. doi:10.1159/000453002.
- Pierrakos C, Smit MR, Pisani L, et al. Lung ultrasound assessment of focal and non-focal lung morphology in patients with acute respiratory distress syndrome. Front Physiol. 2021;12:730857. doi:10.3389/fphys.2021.730857.
- Imai R, Nishimura N, Takahashi O, Tamura T. High-resolution computed tomography for the prediction of mortality in acute respiratory distress syndrome: a retrospective cohort study. Health Sci Rep. 2021;4(4):e418. doi:10.1002/hsr2.418.
- Heo M, Jeong J, Heo I, et al. Association between advanced lung inflammation index and 30-day mortality in patients with acute respiratory distress syndrome. Medicina . 2021;57(8):800. doi:10.3390/medicina57080800.
- Gibson PG, Qin L, Puah SH. COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust. 2020;213(2):54–56.e1. doi:10.5694/mja2.50674.
- Ragab D, Salah EH, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol. 2020;11:1446. doi:10.3389/fimmu.2020.01446.
- Khodadadi H, Salles ÉL, Jarrahi A, et al. Cannabidiol modulates cytokine storm in acute respiratory distress syndrome induced by simulated viral infection using synthetic RNA. Cannabis Cannabinoid Res. 2020;5(3):197–201. doi:10.1089/can.2020.0043.
- Cai N, Jiang M, Wu C, He F. Red cell distribution width at admission predicts the frequency of acute kidney injury and 28-day mortality in patients with acute respiratory distress syndrome. Shock. 2022;57(3):370–377. doi:10.1097/SHK.0000000000001840.
- Sivapalan P, Bonnesen B, Jensen JU. Novel perspectives regarding the pathology, inflammation, and biomarkers of acute respiratory distress syndrome. Int J Mol Sci. 2020;22(1):205. doi:10.3390/ijms22010205.
- Iwata K, Doi A, Ohji G, et al. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med. 2010;49(22):2423–2432. doi:10.2169/internalmedicine.49.4010.
- Lewis SR, Pritchard MW, Thomas CM, Smith AF. Pharmacological agents for adults with acute respiratory distress syndrome. Cochrane Database Syst Rev. 2019;7(7):CD004477. doi:10.1002/14651858.CD004477.pub3.
- Bricher Choque PN, Vieira RP, Ulloa L, et al. The cholinergic drug pyridostigmine alleviates inflammation during LPS-induced acute respiratory distress syndrome. Front Pharmacol. 2021;12:624895. doi:10.3389/fphar.2021.624895.
- Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242(1):186–204. doi:10.1111/j.1600-065X.2011.01033.x.
- Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More than just a barrier: the immune functions of the airway epithelium in asthma pathogenesis. Front Immunol. 2020;11:761. doi:10.3389/fimmu.2020.00761.
- Chua RL, Lukassen S, Trump S, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–979. doi:10.1038/s41587-020-0602-4.
- Erickson NA, Dietert K, Enders J, et al. Soluble mucus component CLCA1 modulates expression of leukotactic cytokines and BPIFA1 in murine alveolar macrophages but not in bone marrow-derived macrophages. Histochem Cell Biol. 2018;149(6):619–633. doi:10.1007/s00418-018-1664-y.
- Nyström EEL, Arike L, Ehrencrona E, Hansson GC, Johansson MEV. Calcium-activated chloride channel regulator 1 (CLCA1) forms non-covalent oligomers in colonic mucus and has mucin 2-processing properties. J Biol Chem. 2019;294(45):17075–17089. doi:10.1074/jbc.RA119.009940.
- Kamada F, Suzuki Y, Shao C, et al. Association of the hCLCA1 gene with childhood and adult asthma. Genes Immun. 2004;5(7):540–547. doi:10.1038/sj.gene.6364124.
- Hegab AE, Sakamoto T, Uchida Y, et al. CLCA1 gene polymorphisms in chronic obstructive pulmonary disease. J Med Genet. 2004;41(3):e27–27. doi:10.1136/jmg.2003.012484.
- Liu CL, Shi GP. Calcium-activated chloride channel regulator 1 (CLCA1): more than a regulator of chloride transport and mucus production. World Allergy Organ J. 2019;12(11):100077. doi:10.1016/j.waojou.2019.100077.
- Centeio R, Ousingsawat J, Schreiber R, Kunzelmann K. CLCA1 regulates airway mucus production and ion secretion through TMEM16A. Int J Mol Sci. 2021;22(10):5133. 3390/ijms22105133. doi:10.3390/ijms22105133.
- Dietert K, Reppe K, Mundhenk L, Witzenrath M, Gruber AD. mCLCA3 modulates IL-17 and CXCL-1 induction and leukocyte recruitment in murine Staphylococcus aureus pneumonia. PLOS One. 2014;9(7):e102606. doi:10.1371/journal.pone.0102606.
- Feng Y, Fang Z, Liu B, Zheng X. p38MAPK plays a pivotal role in the development of acute respiratory distress syndrome. Clinics. 2019;74:e509. doi:10.6061/clinics/2019/e509.
- den Dekker HT, Burrows K, Felix JF, et al. Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J. 2019;53(4):1801795. doi:10.1183/13993003.01795-2018.
- Toda M, Tulic MK, Levitt RC, Hamid Q. A calcium-activated chloride channel (HCLCA1) is strongly related to IL-9 expression and mucus production in bronchial epithelium of patients with asthma. J Allergy Clin Immunol. 2002;109(2):246–250. doi:10.1067/mai.2002.121555.
- Wisnewski AV, Liu J, Redlich CA. Analysis of lung gene expression reveals a role for Cl (-) channels in diisocyanate-induced airway eosinophilia in a mouse model of asthma pathology. Am J Respir Cell Mol Biol. 2020;63(1):25–35. doi:10.1165/rcmb.2019-0400OC.
- Song L, Liu D, Wu C, et al. Antibody to mCLCA3 suppresses symptoms in a mouse model of asthma. PLOS One. 2013;8(12):e82367. doi:10.1371/journal.pone.0082367.
- Hauber H-P, Goldmann T, Vollmer E, et al. LPS-induced mucin expression in human sinus mucosa can be attenuated by hCLCA inhibitors. J Endotoxin Res. 2007;13(2):109–116. doi:10.1177/0968051907079168.
- Ching JC, Lobanova L, Loewen ME. Secreted hCLCA1 is a signaling molecule that activates airway macrophages. PLoS One. 2013;8(12):e83130. doi:10.1371/journal.pone.0083130.
- Cabrita I, Benedetto R, Wanitchakool P, et al. TMEM16A mediates mucus production in human airway epithelial cells. Am J Respir Cell Mol Biol. 2021;64(1):50–58. doi:10.1165/rcmb.2019-0442OC.
- Fang W, Cai S-X, Wang C-L, et al. Modulation of mitogen‑activated protein kinase attenuates sepsis‑induced acute lung injury in acute respiratory distress syndrome rats. Mol Med Rep. 2017;16(6):9652–9658. doi:10.3892/mmr.2017.7811.
- Denham W, Yang J, Wang H, Botchkina G, Tracey KJ, Norman J. Inhibition of p38 mitogen activate kinase attenuates the severity of pancreatitis-induced adult respiratory distress syndrome. Crit Care Med. 2000;28(7):2567–2572. doi:10.1097/00003246-200007000-00064.
- Ipaktchi K, Mattar A, Niederbichler AD, et al. Attenuating burn wound inflammatory signaling reduces systemic inflammation and acute lung injury. J Immunol. 2006;177(11):8065–8071. doi:10.4049/jimmunol.177.11.8065.
- Christie JD, Vaslef S, Chang PK, et al. A randomized dose-escalation study of the safety and anti-inflammatory activity of the p38 mitogen-activated protein kinase inhibitor dilmapimod in severe trauma subjects at risk for acute respiratory distress syndrome. Crit Care Med. 2015;43(9):1859–1869. doi:10.1097/CCM.0000000000001132.
- Alevy YG, Patel AC, Romero AG, et al. IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest. 2012;122(12):4555–4568. doi:10.1172/JCI64896.