
Abstract
Background
Pulmonary emphysema is a condition that causes damage to the lung tissue over time. GBP5, as part of the guanylate-binding protein family, is dysregulated in mouse pulmonary emphysema. However, the role of GBP5 in lung inflammation in ARDS remains unveiled.
Methods
To investigate whether GBP5 regulates lung inflammation and autophagy regulation, the study employed a mouse ARDS model and MLE-12 cell culture. Vector transfection was performed for the genetic manipulation of GBP5. Then, RT-qPCR, WB and IHC staining were conducted to assess its transcriptional and expression levels. Histological features of the lung tissue were observed through HE staining. Moreover, ELISA was conducted to evaluate the secretion of inflammatory cytokines, autophagy was assessed by immunofluorescent staining, and MPO activity was determined using a commercial kit.
Results
Our study revealed that GBP5 expression was altered in mouse ARDS and LPS-induced MLE-12 cell models. Moreover, the suppression of GBP5 reduced lung inflammation induced by LPS in mice. Conversely, overexpression of GBP5 diminished the inhibitory impact of LPS on ARDS during autophagy, leading to increased inflammation. In the cell line of MLE-12, GBP5 exacerbates LPS-induced inflammation by blocking autophagy.
Conclusion
The study suggests that GBP5 facilitates lung inflammation and autophagy regulation. Thus, GBP5 could be a potential therapeutic approach for improving ARDS treatment outcomes, but further research is required to validate these findings.
Introduction
Acute Respiratory Distress Syndrome (ARDS) is a severe respiratory condition affecting millions worldwide. According to recent epidemiological data, ARDS is responsible for approximately 10% of all intensive care unit admissions and is linked to a mortality rate of up to 40%.Citation1,Citation2 Various factors, including pneumonia, sepsis, trauma, and inhalation injury, can cause the disease. The pathophysiology of ARDS involves an overly aggressive inflammatory response occurring in the lungs, leading to alveolar damage and impaired gas exchange. This inflammatory response is characterized by secreting pro-inflammatory cytokines, chemokines, and reactive oxygen species, which can cause tissue damage and impair lung function.Citation3,Citation4 Despite advances in critical care management, the mortality rate of ARDS remains high, and there is currently no specific treatment for the disease. Hence, it is crucial to pinpoint novel therapeutic targets and create efficient treatments for ARDS. In this regard, several studies have shown that inhibiting inflammation can alleviate ARDS.Citation5–7 In this context, pulmonary inflammation has been identified as a key pathological feature of ARDS, and targeting inflammation has been proposed as a potential therapeutic strategy for the disease.
Maintaining cellular homeostasis is crucial, and autophagy is a cellular process that helps achieve this by eliminating damaged organelles and proteins. This process has been found to protect ARDS by eliminating damaged cellular components, preventing the build-up of toxic substances, reducing inflammation, and promoting tissue repair.Citation8,Citation9 Numerous studies have demonstrated the protective role of autophagy in ARDS. One such study found that administering phencyclidine hydrochloride can enhance autophagy in BEAS-2B cells, thereby reducing inflammation and apoptosis associated with ARDS.Citation10 Autophagy is a process of intracellular digestion that is an adaptive response to lung injury caused by exposure to stress agents such as sepsis, hypoxia, and xenobiotics. This injury can manifest as asthma, acute lung injury (ALI), ventilator-induced lung injury (VILI), or pulmonary fibrosis.Citation11
GBP5, also known as Guanylate-binding protein 5, is one of the guanylate-binding proteins. This cluster of proteins involves various cellular signaling, including cell proliferation, responses to inflammation, and cell apoptosis. GBP5 is mainly found in immune cells, particularly in dendritic cells as well as macrophages, and has been found to regulate inflammation. Research has shown that GBP5 can exacerbate rosacea-like skin inflammation by skewing macrophage polarization toward the M1 phenotype through the NF-κB signaling pathway.Citation12 Additionally, studies have found that knocking out GBP5 can improve liver injury and inflammation induced by D-galactosamine/lipopolysaccharide (GalN/LPS). This suggests that GBP5 may have a general pro-inflammatory effect in different cells and diseases. However, no studies have yet found whether GBP5 can regulate the inflammatory response in ARDS.Citation13 Moreover, The GBP5 protein triggers the activation of the NOD-like receptor family, NLRP3 inflammasome responses to specific agents, and its absence impairs host defense and inflammatory responses. This was observed in a study where mice without the GBP5 gene showed defects in IL-1β/IL-18 and caspase-1 cleavage and NLRP3-dependent inflammatory responses.Citation14 Previous research has confirmed that enhancing autophagy can help inhibit the inflammatory response and alleviate ARDS,Citation10,Citation15,Citation16 but the mechanism of GBP5 regulating autophagy function remains unclear.
In the current research, we pursued the role of GBP5 in ARDS and its impact on autophagy function by utilizing the ARDS mouse model and GBP5 gene manipulation in vitro. Our data indicated that high levels of GBP5 can worsen the inflammatory response in ARDS by negatively affecting autophagy function. This suggests that targeting GBP5 could be a potential treatment strategy for ARDS. These findings offer new insights into the underlying mechanisms of pulmonary emphysema and ARDS and could pave the way for new treatments for these conditions. Overall, this study underscores the importance of understanding the molecular mechanisms behind ARDS and the potential benefits of targeting GBP5 as a therapeutic approach to this life-threatening disease.
Methods and materials
Animal procedure and ARDS mouse model
C57BL/6 mice aged 8–12 week were purchased from Jackson Laboratory (Cat#: 000664). Animal attending was performed following the guidelines of the Animal Committee of Kangtai medical inspection service Hebei Co., LTD (Ethical Approval NO. MDL2022-12-12-03). The study received approval from "The Chinese Board for Animal Experiments." Animals were weighed daily and removed from the study if their body weight dropped by 20% or more from the baseline or if there was a weight loss of over 10% between measurements. In addition, animals were eliminated from the study if they displayed severe dehydration, lack of movement, skin lesions, continuous tremors, or respiratory failure. Throughout the study, animals had unlimited access to food and water.
To establish the mouse model of ARDS, this study used Lipopolysaccharides from Escherichia coli O111:B4 (LPS, Cat. No. L2630, Sigma-Aldrich) stimulation to induce ARDS mouse model, using a previously established protocol.Citation17 The mice were randomly separated into the LPS group and the control group. The LPS group was given an intraperitoneal injection of LPS (5 mg/kg) to trigger ARDS, whereas the control group was administered saline. After 24 h, lung tissues were gathered for further examination. The animals were kept in a pathogen-free environment with a 12-h light/dark cycle and given standard rodent chow and water ad libitum.
Cell culture and treatment
Here, we constructed an ARDS cell model to mimic aspects of the ARDS environment at the cellular level. Briefly, MLE-12 cells (ATCC, Shanghai, China) were grown in DMEM/F12 medium with 10% FBS and 1% penicillin/streptomycin. The cells were seeded in 6-well plates and left to adhere for 24 h. Fresh medium was added every 2–3 days. After attaching, the cells were treated with LPS at a concentration of 1 μg/mL for 24 h. All of this was done in a humidified atmosphere of 5% CO2 at 37 °C.Citation18 To maintain optimal growth, cells were passaged once they reached 80-90% confluence. 0.25% trypsin-EDTA (T4049, Sigma-Aldrich) was used for cell dissociation. In , a concentration of 1uM Rapamycin (Cat. No. 1292, Tocris) was included.
Plasmid transfection for knockdown and overexpression of GBP5
To unveil the molecular mechanism of GBP5 in the pathogenesis of ARDS, we performed knockdown and overexpression experiments in MLE-12 cells using plasmid transfection. Specifically, we designed and constructed sh-NC (negative control), sh-GBP5, and OV-GBP5 (overexpression of GBP5) plasmids customized from Addgene, respectively. The sequences of sh-GBP5 are as follows: Sense: 5′-CAAGCTGACCCTGAAGTTCAT-3′, Anti-sense: 5’GCTCGGCTTTACTTAAGGATA-3′. RT-qPCR and WB analysis confirmed the knockdown efficiency of sh-GBP5, as well as the overexpression efficiency of OV-GBP5. According to the manufacturer’s instructions, we transfected the plasmids into MLE-12 cells using Lipofectamine 3000 (L3000001, Thermofisher).
RT-qPCR
To obtain RNA from MLE-12 cells, TRIzol reagent (T9424, Sigma-Aldrich) was used per the manufacturer’s instructions. The concentration of RNA was assessed using a NanoDrop spectrophotometer (2000 series, Thermo Scientific). Samples with an A260/A280 ratio ranging from 1.8 to 2.0 were used for further analysis. PrimeScript RT reagent kit was used to synthesize cDNA from 1 μg of total RNA, which was then diluted with nuclease-free water. The expression levels of GBP5 and other target genes were determined using RT-qPCR, the SYBR Green PCR Master Mix (A46109, ThermoFisher), and the ABI 7500 Real-Time PCR System (4351107, Applied Biosystems™). The PCR conditions were as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The specificity of the PCR products was confirmed by melting curve analysis. The 2^-ΔΔCt method was used to calculate the relative expression levels of the target genes, with GAPDH as the internal control. All experiments were conducted in triplicate, and statistical methods were used to analyze the results. The primer sequences for RT-qPCR were listed in .
Table 1. Primer sequences for qRT-PCR.
Western blot (WB)
After extracting total protein from the cells, we separated equal amounts using SDS-PAGE and transferred them onto PVDF membranes (GVWP02500, Sigma-Aldrich). After blocking the membranes with 5% nonfat milk in TBST buffer, we incubated them overnight at 4 °C with primary antibodies against GBP5 (ab96119, Abcam). We then washed the membranes with TBST buffer and incubated them with HRP-conjugated secondary antibodies for an hour at room temperature. Using an ECL detection system, we visualized the protein bands and quantified them using ImageJ software.
Histopathological staining
The lung tissue samples were preserved using 4% paraformaldehyde and embedded in paraffin blocks. To carry out HE and IHC staining, 4-μm-thick sections of the blocks were cut. The tissue slices were then deparaffinized and rehydrated with xylene and graded ethanol solutions. Hematoxylin was applied (H3136, Sigma-Aldrich) for 5 min, and the sections were washed with distilled water. Eosin was then applied (45260, Sigma-Aldrich) for 2 min and the sections were washed with distilled water. The sections were cleared in a 1% HCl solution in 70% alcohol. For IHC staining, paraffin lung sections were dewaxed with xylene and rehydrated with ethanol. Endogenous peroxidase activity was blocked by incubation with 0.3% hydrogen peroxide. The sections were then probed with primary antibodies against GBP5 (ab96119, Abcam) or CD68 (sc-7084, Santa Cruz Biotechnology) overnight and then incubated with a horseradish peroxidase-conjugated secondary antibody for 0.5 h. Finally, the sections were mounted using a coverslip and mounting medium. The histological features of the lung tissue and protein expression of GBP5 and CD68 were analyzed under a light microscope.
Immunofluorescent staining
To prepare tissue samples, we used 4% PFA. Afterward, we embedded the samples at −80 °C. Using a cryostat, we cut sections of 6 μm thickness, which we mounted on a glass slide. MLE-12 cells were also fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100. Both the slices and cells were then subjected to incubation with primary antibody against LC3B (ab192890, Abcam) overnight at 4 °C. Subsequently, we incubated them with secondary antibodies conjugated with fluorescent dyes (Goat Anti-Rabbit IgG H&L, ab150077, Abcam) for 60 min in the dark at room temperature. After washing the sections with PBS, we mounted them with a coverslip using a mounting medium. Finally, we used a fluorescent microscope (OLYMPUS, IX71) to photograph the samples.
Mmeasurement of myeloperoxidase (MPO) activity
Determination of MPO was conducted by previous protocols.Citation19,Citation20 To achieve this, lung samples were homogenized and then centrifuged at 12,000 g for 20 min at 4 °C. The supernatants were collected and used in an MPO Detection Kit from Nanjing Jiancheng Bioengineering Institute in Nanjing, China. The assay was conducted by measuring absorbance at 460 nm using a plate reader after adding the supernatants to a 96-well plate. MPO activity was presented as U/g of total protein, which was calculated using the BCA protein assay kit.
ELISA
The levels of inflammatory cytokines TNF-α and IL-1β in BALF were detected using the ELISA assay, as per the manufacturer’s instructions. A microplate reader was used to measure the absorbance at 450 nm. The levels of TNF-α and IL-1β were calculated based on standard curves generated with known concentrations of recombinant cytokines. To collect lung tissues and BALF from mice, CO2 inhalation was used to euthanize the animals. The chest cavity was opened, and the lungs were perfused with 1 mL of PBS through the right ventricle to remove blood. Using a tissue homogenizer, the left lung was homogenized in 1 mL of PBS, and the homogenates were centrifuged at 12,000 g for 10 min at 4 °C. The supernatants obtained were collected and stored at −80 °C for further analysis.
The trachea was exposed and cannulated with a 20-gauge catheter for BALF collection. The lungs were then lavaged three times with 0.5 mL of PBS, and the recovered BALF was centrifuged at 500 g for 10 min at 4 °C. The supernatants obtained were collected and stored at −80 °C for further analysis. The ELISA assay was used to determine TNF-α, IL-1β, and IL-6 levels in the lung tissues and BALF.
Statistical analysis
The statistical analysis for this research was conducted on SPSS version 25.0 by IBM Corp. in Armonk, NY, USA. The data is presented as mean ± standard deviation (SD). To compare the means of multiple groups, we used one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. For comparing the standards of two groups, a simple student t-test was used. In order to assess the significance of differences between the experimental groups, a p-value of less than 0.05 was used as the threshold for statistical significance.
Results
Upregulation of GBP5 in LPS-induced mouse model and at the cellular level
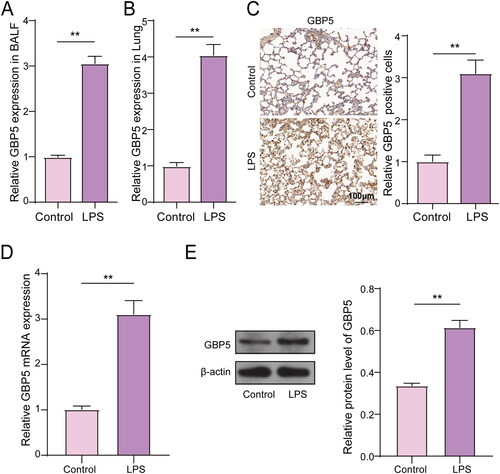
According to prior studies regarding GBP5 regulating autophagy,Citation14 we predict that GBP5 expression may be altered in response to various inflammatory stimuli during ARDS. GBP5 may control immune responses and inflammation in the lung during ARDS. To investigate the expression of GBP5 in mouse ARDS, we used an LPS-induced mouse model (n = 5) and collected lung tissue and BALF for analysis. The mice were equally grouped into Control and LPS groups. We conducted RT-qPCR to measure the mRNA levels of GBP5 in both lung tissue and BALF. Our results showed that in BALF, the mRNA level of GBP5 in the LPS group was seven times higher than in the control group (). In lung tissue, GBP5 expression increased by four times in the LPS group (). We also performed IHC to detect GBP5 in lung tissue and found it was significantly upregulated in ARDS lung tissue ( C). To further investigate GBP5 expression in ARDS cells, we used LPS-stimulated MLE-12 cells to create an ARDS cell model. RT-qPCR and WB were used to measure GBP5 expression and it found that both mRNA and protein levels of GBP5 were elevated in the LPS group (). Collectively, these findings indicate an upregulation of GBP5 in response to LPS-induced inflammation in both a mouse model and cell culture of ARDS, suggesting its potentially pivotal role in mediating the immune response and inflammation during ARDS.
Figure 1. GBP5 Expression was altered in mouse ARDS models.
(A) Bar graphs showing population data of GBP5 mRNA levels in LPS and control groups from lung tissue of ARDS mice model (n = 5). (B) Same as (A), but in BALF. (C) Example images of IHC staining for GBP5 in lung tissue. (D) Population data of GBP5 mRNA levels in an ARDS cell model established by LPS-induced MLE-12 cell lines. (E) Same as (D), but in protein levels. Structures represent mean ± SD. The asterisk indicates p < 0.05.
Knockdown of GBP5 inhibits LPS-induced lung inflammation in mice
Considering our earlier discovery that GBP5 expression is altered in mouse models of ARDS, we sought to explore the potential therapeutic benefits of modulating GBP5 expression in ARDS. We employed gene manipulation and gene editing techniques to increase and decrease GBP5 expression and observed the effects on ARDS. Based on our earlier findings, we predicted that reducing GBP5 levels could inhibit LPS-induced lung inflammation in mice. We used an LPS-induced mouse model of ARDS (n = 5) and collected lung tissue and BALF for analysis. We examined the role of GBP5 in the development of ARDS by using shRNA to reduce GBP5 expression in the lung tissue of LPS-treated mice. Our results revealed that shRNA-mediated reduction of GBP5 in lung tissue significantly inhibited LPS-induced upregulation of GBP5 expression. Specifically, the mRNA levels of GBP5 in the sh-GBP5 group returned to normal levels, similar to those in the control group without LPS treatment, and were three times lower than those in the LPS + sh-NC group ().
Figure 2. Reducing GBP5 levels inhibited lung inflammation induced by LPS in mice.
(A) Bar graph showing population data of GBP5 levels in BALF for the Control, LPS + sh-NC, and LPS + sh-GBP5 groups. n = 5 mice. (B) HE staining images showing pathological changes in lung tissue. (C) Bar graph showing the results of MPO activity assay. (D) Representative images of IHC staining for CD68 in lung tissue. (E) Spread dot plot showing ELISA measurements of inflammatory factors TNF-α and IL-1β in BALF. Structures represent mean ± SD. ** p < 0.01, *** p < 0.001.
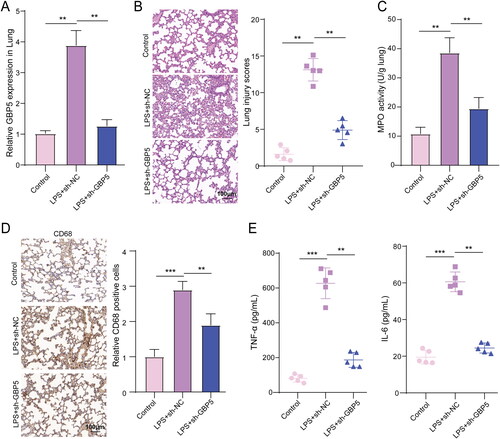
Additionally, LPS treatment caused significant pathological changes in lung tissue, including damage to lung structure, thickening of alveolar septa, infiltration of inflammatory cells, hemorrhage in alveoli, and edema in interstitial and alveolar spaces. Reducing GBP5 levels significantly inhibited LPS-induced pathological changes in lung tissue, as demonstrated by population data showing a 2.68-fold decrease in the sh-GBP5 group compared to the LPS + sh-NC group (). We also measured MPO activity to assess the activation and accumulation of neutrophils in lung tissue. Our findings indicate that the activity of MPO was increased significantly by LPS, whereas inhibiting GBP5 levels resulted in a significant reduction of LPS-induced MPO activity (), which was similar to the variation trending GBP5 mRNA levels observed in . Additionally, reducing GBP5 levels significantly inhibited LPS-induced expression of CD68 in lung tissue, a macrophage activation marker (). Furthermore, reducing GBP5 levels significantly inhibited the secretion of inflammatory cytokines (TNF-α and IL-1β) in BALF, which was decreased by 3.36-fold and 2.48-fold compared to the LPS + sh-NC group, respectively (). To date, our findings indicate that GBP5 plays a crucial role in the development of ARDS by regulating inflammatory responses and immune cell activation, reducing GBP5 levels may reverse pathological and cellular damage derived from ARDS.
Downregulation of GBP5 enhances autophagy inhibited by LPS in ARDS mice
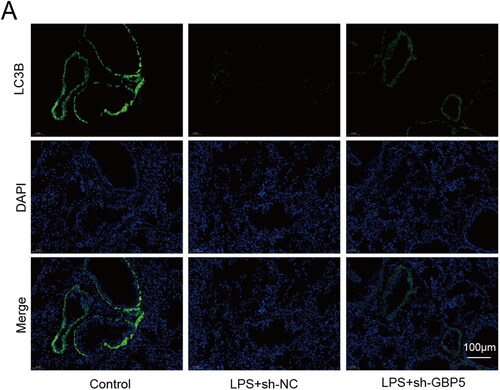
As our findings revealed that excessive GBP5 facilitated the pathogenesis of ARDS, and autophagy is involved in regulating inflammation and tissue repair in ARDS,Citation9,Citation10 we aimed to investigate the potential connection between GBP5 and autophagy. To this end, we used a mouse model of ARDS and collected lung tissue from three groups of mice: Control, LPS + sh-NC, and LPS + sh-GBP5. We utilized immunofluorescence staining to detect the expression of LC3B, a widely used autophagy marker that reflects autophagosome formation and the extent of autophagy activity.Citation21,Citation22 Our results showed that the fluorescence signal of LC3B was significantly increased in the LPS + sh-GBP5 group compared to the LPS + sh-NC group, indicating that knockdown of GBP5 may enhance autophagy in the lung tissue of LPS-treated mice (). The upregulation of LC3B in response to knockdown of GBP5 is consistent with previous studies that have shown LC3B to be a reliable marker of autophagy activity in various cell types and tissues.Citation23
Figure 3. Decreasing GBP5 expression increased autophagy that LPS suppressed in ARDS mice.
(A) Representative immunofluorescence images showing LC3B for the Control, LPS + sh-NC, and LPS + sh-GBP5 groups.
GBP5 exacerbates inflammation in LPS-treated MLE-12 cells by inhibiting autophagy
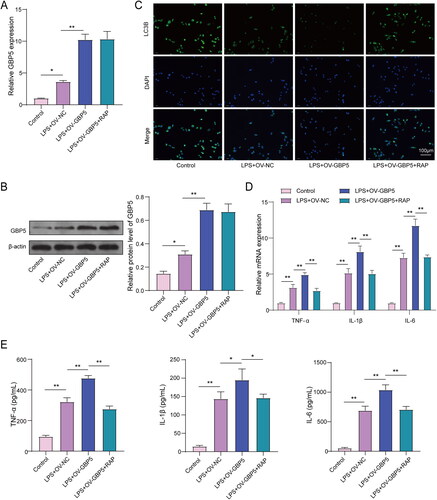
In our continued investigation into the role of GBP5 in ARDS, we aimed to examine the effects of increasing GBP5 expression in an ARDS cell model. We divided the cells into four groups: Control, LPS + OV-NC (overexpression of negative control), LPS + OV-GBP5 (overexpression of GBP5), and LPS + OV-GBP5 + RAP (overexpression of GBP5 with rapamycin treatment, an autophagy activator). We first performed RT-qPCR and WB assay to measure GBP5 expression. Our results revealed that overexpressing GBP5 further increased the high expression of GBP5 induced by LPS in MLE-12 cells. The addition of rapamycin (RAP), an autophagy activator, did not affect GBP5 expression. In the OV-GBP5 and OV-GBP5-RAP groups, GBP5 expression was 15 times upregulated, but there was no difference between the OV-GBP5 and OV-GBP5-RAP groups (), indicating that RAP did not further alter GBP5 expression. Additionally, we performed immunofluorescence to detect LC3B, a marker of autophagy activity. showed that LC3B signal fluorescence intensity was slightly enhanced in the control group than in the LPS + OV-GBP5 + RAP group, significantly pronounced than in the LPS + OV-NC group, and lowest in the LPS + OV-GBP5 group. However, RAP reversed the inhibitory effect of OV-GBP5 on LC3B. Furthermore, we found that overexpressing GBP5 increased the secretion of inflammatory factors induced by LPS. However, The pro-inflammatory effect of OV-GBP5 was reversed by RAP, resulting in a decrease in the secretion of inflammatory factors back to the level observed in the NC group. This decrease amounted to approximately 1.5 times (). These findings suggest that increasing GBP5 may promote the secretion of inflammatory factors and that RAP can reverse this effect by activating autophagy without changing the transcription level of GBP5.
Figure 4. GBP5 Increased inflammation in LPS-stimulated MLE-12 cells by suppressing autophagy.
ARDS cell model established by LPS-induced MLE-12 cell lines (n = 3). (A) Bar graphs showing RT-qPCR measurements of GBP5 mRNA levels in LPS-stimulated MLE-12 cells used to create an ARDS cell model. Groups: Control, LPS + OV-NC, LPS + OV-GBP5, LPS + OV-GBP5 + RAP. (B) Same as (A), but showing protein levels from WB. (C) Graph showing population data of immunofluorescence detection of autophagy-related protein LC3B. (D-E) Population data of RT-qPCR and ELISA measurements of TNF-α, IL-1β, and IL-6. Structures represent mean ± SD.* p < 0.05, ** p < 0.01.
Discussion
In this study, high levels of GBP5 can exacerbate the inflammatory response in ARDS by negatively affecting autophagy function. A possible treatment approach for ARDS could be to target GBP5, as it may improve autophagy function and reduce inflammation.
ARDS is a critical condition marked by severe inflammation and fluid buildup in the lungs, resulting in respiratory failure and high mortality rates. Despite progress in critical care medicine, managing ARDS remains challenging, and new treatment approaches are needed. Understanding the pathophysiology of ARDS is a multifaceted process that involves various mechanisms like inflammation, oxidative stress, endothelial dysfunction, and impaired clearance of alveolar fluid.Citation24 The inflammatory response in ARDS is characterized by the release of cytokines such as TNF-α, IL-1β, and IL-6, which can damage lung tissue and impair gas exchange.Citation25 In recent years, our understanding of the pathophysiology of ARDS has improved significantly, resulting in new treatment approaches. For instance, prone positioning, which involves placing patients face down to improve oxygenation, has decreased mortality in patients with severe ARDS.Citation26 Additionally, extracorporeal membrane oxygenation (ECMO), which provides temporary support for failing lungs, has also been shown to improve outcomes in patients with severe ARDS.Citation27
Despite these advances, much remains unknown about the pathophysiology of ARDS, and further research is needed to identify new treatment approaches. One area of research that has gained attention recently is the role of autophagy in ARDS. Maintaining cellular balance and eliminating damaged organelles and proteins is a vital function of autophagy, a crucial cellular process. If this process is not regulated properly, it can lead to the development of various illnesses, including ARDS.Citation28 Recent findings have indicated that enhancing autophagy can reduce inflammation and improve outcomes in animal models of ARDS.Citation29,Citation30 In this study, we examined the role of GBP5 in ARDS and its impact on autophagy function.
GBP5 belongs to the GBP family, and it serves as an essential part of the innate immune response to viral and bacterial infections.Citation31 Recent studies have suggested that GBP5 regulates inflammation in various diseases, such as sepsis, rheumatoid arthritis, and cancer.Citation32,Citation33 In our study, we examined how GBP5 in ARDS impacts autophagy function. There are conflicting results in the research on the part of GBP5 in regulating autophagy and inflammation. While some studies have suggested that GBP5 can promote promoting inflammation 14, 37, 38, another study has indicated that GBP5 can inhibit inflammation by promoting autophagy-mediated degradation 31. These findings suggest that the role of GBP5 in regulating autophagy and inflammation is complex and requires further investigation.
Our results indicated that high levels of GBP5 can exacerbate the inflammatory response in ARDS by negatively affecting autophagy function (). This aligns with previous research findings that GBP5 can promote inflammation by activating the NF-κB signaling pathway and increasing the secretion of cytokines such as TNF-α and IL-6.Citation34 Additionally, GBP5 is upregulated in various inflammatory diseases.Citation13,Citation35 Herein, our results showed that GBP5 interacts with autophagy-related pathways, leading to inhibition of autophagy, as evidenced by the decreased expression of LC3B, a marker of autophagosome formation, upon GBP5 overexpression. Conversely, the knockdown of GBP5 enhances autophagy, as indicated by increased LC3B expression. The molecular mechanism through which GBP5 inhibits autophagy likely involves interactions with components of the autophagy machinery, although the precise interactions remain to be fully elucidated. Collectively, our findings support GBP5 as a potential therapeutic target for ARDS by modulating autophagy to alleviate inflammation. Previous work also supports this by showing that targeting GBP5 can reduce inflammation and improve outcomes in animal models 33. However, additional research is needed to fully elucidate the specific molecular interactions via which GBP5 influences autophagy pathways.
The process of autophagy is crucial in keeping cellular balance and has been proven to possess anti-inflammatory properties. Several studies have shown that autophagy can suppress inflammation by removing damaged organelles and proteins, thereby preventing the activation of inflammatory pathways.Citation36,Citation37 While research on the role of GBP5 in regulating autophagy is limited, recent studies have suggested that GBP5 may mediate autophagy-mediated inflammation. For instance, a study showed that GBP5 facilitates the assembly and activation of the NLRP3 inflammasome through interactions with NLRP3Citation27. Since the NLRP3 inflammasome is a crucial mediator of inflammation, these findings suggest that GBP5 may regulate inflammation through its effects on autophagy. This study indicates that inhibition of GBP5 may control inflammation by promoting the clearance of pro-inflammatory cytokines through activating autophagy.
While the study has shown encouraging outcomes, it is crucial to acknowledge that some limitations need attention and resolution. For one, the research was conducted on a mouse model, and it is still being determined whether the findings can be applied to humans. Additionally, clinical data is necessary to allow our understanding of how these results relate to human disease. Moreover, the long-term effects of manipulating GBP5 were not examined, and future research is needed to determine if the observed impacts are sustainable. Lastly, while this study focused on the role of GBP5 in regulating lung inflammation and autophagy, its potential involvement in other aspects of ARDS pathogenesis warrants further investigation. In summary, while these findings are encouraging, additional research is necessary to validate and expand upon them.
Conclusion
This study presents compelling evidence that the protein GBP5 significantly regulates lung inflammation and autophagy in ARDS. The results suggest that targeting GBP5 could be a potential therapeutic strategy for treating ARDS and provide new insights into the pathogenesis of pulmonary emphysema and ARDS. Overall, this study highlights the potential of targeting GBP5 as a novel therapeutic approach for ARDS and provides a foundation for further research in this area.
Ethical approval
Animal attending was performed following the guidelines of the Animal Committee of The First People’s Hospital of Chenzhou.
Acknowledgment
The authors would like to thank all the participants who contributed to this study. We also thank the technical support provided by the laboratory staff.
Disclosure statement
The authors declare no conflict of interest.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Additional information
Funding
References
- Máca J, Jor O, Holub M, et al. Past and present ARDS mortality rates: a systematic review. Respir Care. 2017;62(1):113–122. doi:10.4187/respcare.04716.
- Welker C, Huang J, Gil IJN, Ramakrishna H. 2021 acute respiratory distress syndrome update, with coronavirus disease 2019 focus. J Cardiothorac Vasc Anesth. 2022;36(4):1188–1195. doi:10.1053/j.jvca.2021.02.053.
- Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204–7218. doi:10.18632/oncotarget.23208.
- Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023–17023. doi:10.1038/sigtrans.2017.23.
- Liu B, Cheng Y, Wu Y, et al. Emodin improves alveolar hypercoagulation and inhibits pulmonary inflammation in LPS-provoked ARDS in mice via NF-kappaB inactivation. Int Immunopharmacol. 2020;88:107020. doi:10.1016/j.intimp.2020.107020.
- Sears B. Anti-inflammatory diets. J Am Coll Nutr. 2015;34(sup1):14–21. doi:10.1080/07315724.2015.1080105.
- Tung YT, Wei CH, Yen CC, et al. Aspirin attenuates hyperoxia-induced acute respiratory distress syndrome (ARDS) by suppressing pulmonary inflammation via the NF-kappaB signaling pathway. Front Pharmacol. 2021;12:793107. doi:10.3389/fphar.2021.793107.
- Qu M, Chen Z, Qiu Z, et al. Neutrophil extracellular traps-triggered impaired autophagic flux via METTL3 underlies sepsis-associated acute lung injury. Cell Death Discov. 2022;8(1):375. doi:10.1038/s41420-022-01166-3.
- Zhao H, Chen H, Xiaoyin M, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation. 2019;42(2):426–439. doi:10.1007/s10753-018-00952-5.
- Liu H, Wang S, Gong L, et al. SIRT6 ameliorates LPS-induced apoptosis and tight junction injury in ARDS through the ERK1/2 pathway and autophagy. Int J Med Sci. 2023;20(5):581–594. doi:10.7150/ijms.80920.
- Xie Y, Hu W, Chen X, et al. Identification and validation of autophagy-related genes in exogenous sepsis-induced acute respiratory distress syndrome. Immun Inflamm Dis. 2022;10(10):e691. doi:10.1002/iid3.691.
- Zhou L, Zhao H, Zhao H, et al. GBP5 exacerbates rosacea-like skin inflammation by skewing macrophage polarization towards M1 phenotype through the NF-kappaB signalling pathway. J Eur Acad Dermatol Venereol. 2023;37(4):796–809. doi:10.1111/jdv.18725.
- Ding K, Li X, Ren X, et al. GBP5 promotes liver injury and inflammation by inducing hepatocyte apoptosis. Faseb J. 2022;36(1):e22119. doi:10.1096/fj.202101448R.
- Shenoy AR, Wellington DA, Kumar P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336(6080):481–485. doi:10.1126/science.1217141.
- Chichger H, Rounds S, Harrington EO. Endosomes and autophagy: regulators of pulmonary endothelial cell homeostasis in health and disease. Antioxid Redox Signal. 2019;31(13):994–1008. doi:10.1089/ars.2019.7817.
- Liu X, Gao C, Wang Y, et al. BMSC-derived exosomes ameliorate LPS-induced acute lung injury by miR-384-5p-controlled alveolar macrophage autophagy. Oxid Med Cell Longev. 2021;2021:9973457. 20219973457. doi:10.1155/2021/9973457.
- Kaspi H, Semo J, Abramov N, et al. MSC-NTF (NurOwn(R)) exosomes: a novel therapeutic modality in the mouse LPS-induced ARDS model. Stem Cell Res Ther. 2021;12(1):72. doi:10.1186/s13287-021-02143-w.
- Chimenti L, Morales-Quinteros L, Puig F, et al. Comparison of direct and indirect models of early induced acute lung injury. Intensive Care Med Exp. 2020;8(Suppl 1):62. doi:10.1186/s40635-020-00350-y.
- Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19(1):96. doi:10.1186/s12967-021-02745-1.
- Wang QL, Yang L, Liu ZL, et al. Sirtuin 6 regulates macrophage polarization to alleviate sepsis-induced acute respiratory distress syndrome via dual mechanisms dependent on and independent of autophagy. Cytotherapy. 2022;24(2):149–160. doi:10.1016/j.jcyt.2021.09.001.
- Satyavarapu EM, Das R, Mandal C, et al. Autophagy-independent induction of LC3B through oxidative stress reveals its non-canonical role in anoikis of ovarian cancer cells. Cell Death Dis. 2018;9(10):934. doi:10.1038/s41419-018-0989-8.
- Schmitz KJ, Ademi C, Bertram S, et al. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J Surg Oncol. 2016;14(1):189. doi:10.1186/s12957-016-0946-x.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356.
- Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0.
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi:10.1056/NEJM200005043421806.
- Guérin C, Reignier J, Richard J-C, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368(23):2159–2168. doi:10.1056/NEJMoa1214103.
- Combes A, Hajage D, Capellier G, et al. Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. N Engl J Med. 2018;378(21):1965–1975. doi:10.1056/NEJMoa1800385.
- Su Z, Yang Z, Xu Y, et al. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48. doi:10.1186/s12943-015-0321-5.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi:10.1038/nri3532.
- Lopez-Soler RI, Nikouee A, Kim M, et al. Beclin-1 dependent autophagy improves renal outcomes following unilateral ureteral obstruction (UUO) injury. Front Immunol. 2023;14:1104652. doi:10.3389/fimmu.2023.1104652.
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5(2):133–147. doi:10.1038/nrm1313.
- Haque M, Singh AK, Ouseph MM, Ahmed S. Regulation of synovial inflammation and tissue destruction by guanylate binding protein 5 in synovial fibroblasts from patients with rheumatoid arthritis and rats with adjuvant-induced arthritis. Arthritis Rheumatol. 2021;73(6):943–954. doi:10.1002/art.41611.
- Yu X, Jin J, Zheng Y, et al. GBP5 drives malignancy of glioblastoma via the Src/ERK1/2/MMP3 pathway. Cell Death Dis. 2021;12(2):203. doi:10.1038/s41419-021-03492-3.
- Kim BH, Shenoy AR, Kumar P, et al. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332(6030):717–721. doi:10.1126/science.1201711.
- Kuenz B, Lutterotti A, Ehling R, et al. Cerebrospinal fluid B cells correlate with early brain inflammation in multiple sclerosis. PLoS One. 2008;3(7):e2559. doi:10.1371/journal.pone.0002559.
- Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and inflammation. Annu Rev Immunol. 2018;36:73–101. doi:10.1146/annurev-immunol-042617-053253.
- White E, Karp C, Strohecker AM, et al. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22(2):212–217. doi:10.1016/j.ceb.2009.12.008.