Abstract
The goal of this review is to provide a brief introduction to the effects of hyperthermia on cellular structures and physiology. The review focuses on the effects of hyperthermia thought to contribute to the enhancement of cancer therapy namely the mechanisms of cell killing and the sensitization of cells to ionizing radiation or chemotherapeutic agents. Specifically the review addresses four topics: hyperthermia induced cell killing, mathematical models of cell killing, mechanisms of thermal effects in the hyperthermia temperature range and effects on proteins that contribute to resistance to other stresses, i.e., DNA damage. Hyperthermia has significant effects on proteins including unfolding, exposing hydrophobic groups, and aggregation with proteins not directly altered by hyperthermia. Protein aggregation has effects throughout the cell but has a significant impact within the nucleus. Changes in the associations of nuclear proteins particularly those involved in DNA replication cause the stalling of DNA replication forks and lead to the induction of DNA damage such as double strand breaks. It has long been recognized that heat has effects on plasma membrane protein distribution alters the permeability of plasma membranes resulting in a calcium spike and disrupts the mitochondrial membrane potential resulting in the change in the redox status of cells. These effects contribute to the protein unfolding effects of hyperthermia and contribute to effects observed in the nucleus. Thus heat effects on multiple cellular targets can be integrated through global effects on protein folding to affect specific end points such as cell killing and sensitization to additional stresses.
Introduction
The goal of this review is to introduce the reader to the effects of hyperthermia on cellular structures and physiology. Since this field is too broad for a 30-minute talk or a review article of modest length, I have chosen to highlight several aspects of the effects of hyperthermia, focusing on those that contribute to the enhancement of cancer therapy, namely the mechanisms of cell killing and the sensitization of cells to ionizing radiation or chemotherapeutic agents. The potential importance of this subject for cancer treatment has been recently highlighted by Coffey et al. Citation[1]. Specifically, the review addresses four topics: (1) hyperthermia induced cell killing, (2) mathematical models of cell killing, (3) mechanisms of thermal effects in the hyperthermia temperature range and (4) effects on proteins that contribute to resistance to other stresses, for example, DNA damage.
Cell killing
It has been long recognized that hyperthermia in the 40–47°C temperature range kills cells in a reproducible time and temperature dependent manner. Survival curves for temperatures in the 43–47°C range typically show a shoulder with an exponential reduction in clonogenic survival as a function of time at a given temperature Citation[2–4]. In contrast, cell survival curves for temperatures of 42.5°C and below, depending on the cell line, will show a shoulder, an exponential portion of cell killing followed by a plateau in cell killing, due to the development of chronic thermal tolerance. The principal conclusion from these studies is that for hyperthermia, thermal dose is a combination of time and temperature.
Mathematical models of cell killing
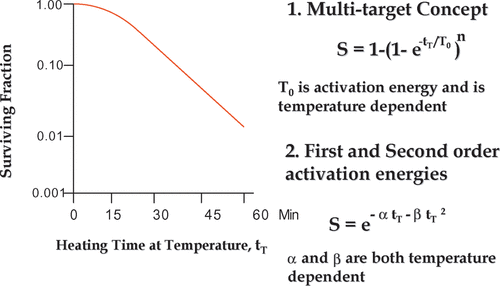
Mathematical representation of survival curves can be written by several mathematical treatments including mathematical models. Such models were based on survival curves from radiation. The earliest such equation was based on the multi-target single hit model, resulting in the survival equation: S = 1 − (1 − etT/To)n, where tT is time at temperature T and To is the activation energy for cell killing at temperature T Citation[5]. In this model the To parameter is temperature dependent, and is related to the reciprocal of the absolute temperature by Arrhenius analysis Citation[5]. On the other hand, the parameter, n, did not follow the same function with absolute temperature for all cell lines Citation[6]. An alternate approach was to use a linear quadratic type equation where S = exp (−αtT − βtT²). As above, tT is the time at a given temperature T, while α and β are arbitrary constants. However, α is linear function of the reciprocal of the absolute temperature and β is a linear function of the reciprocal of the absolute temperature squared, according to the Arrhenius analysis Citation[4], Citation[7]. Another model, developed by Mackey et al. Citation[8] utilized the distribution of an arbitrary temperature dependent parameter, which needed to be above a specified threshold for the cell to be clonogenic. This model was able to simulate the development of chronic thermal tolerance and the hypersensitivity of S phase cells, two important aspects of cell killing that were not addressed by the two earlier models. While the Mackey model was based on thermodynamic considerations, evidence for a direct link between the empirical parameters and thermodynamic constants remained elusive. Thus, a cell survival model, completely determined by thermodynamics and the biological response to heat induced changes in protein folding has not been derived from the first principles and is still a tool that is lacking for the field of hyperthermia. Such a model would be important for any improved definition and quantification of thermal dose.
Mechanisms of thermal effects in the hyperthermia temperature range
In the hyperthermia temperature range (40–47°C), the main effects at the cellular level that are of relevance to cancer therapy are cell killing and those effects that alter the resistance of cells to radiation and/or chemotherapeutic agents. While it is acknowledged that at the tissue, organ and whole body level there are numerous effects that can contribute to the clinical uses of hyperthermia; this review focuses on effects at the cellular level. Thus, we will discuss cell killing and those effects that alter resistance to other cellular stresses.
Prior to discussing the mechanisms of cell death it is important to consider the types of cell death that are induced by hyperthermia. The types of cell death induced by hyperthermia include heat induced apoptosis Citation[9], mitotic catastrophe secondary to alterations in the proteins that support DNA metabolism Citation[10–11]. The type of cell death induced by heat shock is highly cell-type and temperature dependent Citation[12]. In fact, most cell lines die of a combination of death processes.
The goal of hyperthermia research is to find the molecular mechanisms by which heat kills tumour cells or any cells and the mechanisms by which hyperthermia radiosensitizes cells to radiation or chemotherapeutic agents. A major problem is that hyperthermia causes a large number of macromolecular changes and affects functions in all cellular compartments at temperatures above 43°C. Although significantly fewer macromolecular changes occur in the 40.5° to 42°C range, these changes are still numerous and occur in multiple cellular compartments. The challenge is to determine which molecular changes are critical for the relevant endpoint such as tumour cell killing and sensitization to chemo and radiation therapy. By the late 1980s there was a significant body of literature describing heat effects on DNA, proteins, lipids and other cellular components Citation[13]. Information on the effects of hyperthermia on cellular macromolecules has been growing rapidly since that time, making a comprehensive review almost impossible. However, no clear, detailed mechanism for cell killing or sensitization to other stresses has been established. Nevertheless, the main consensus that has emerged is that the mechanism by which hyperthermia kills cells and/or sensitizes cells to radiation involves the unfolding and subsequent aggregation of proteins Citation[14]. Therefore, one approach would be to focus on how heat affects those proteins that contribute to resistance to other stresses.
Effects on proteins that contribute to resistance to other stresses, for example, DNA damage
Hyperthermia effects on proteins include unfolding and exposing hydrophobic groups, and aggregation of unfolded proteins due to the interactions of the exposed hydrophobic groups (, panel A). Aggregation of proteins can include proteins not directly altered by hyperthermia, thereby affecting a larger number of proteins than those directly altered by heat shock. Protein aggregation and unfolding will have effects throughout the cell but should have significant impact within the nucleus because of the large number of proteins and the large amount of DNA packed within it. For example, over 2 m of DNA (5 × 109 base pairs) and a protein mass within the nucleus is from 3 to 6 times that of the mass of DNA packed within the nucleus Citation[15–20]. The second reason that protein unfolding within the nucleus can have a significant impact is that the proteins within the nucleus are critical to managing the proper transcription, replication and repair of DNA Citation[17–20]. Therefore disruptions of these proteins and their function(s) should have critical consequences. Specifically, disruption of the proteins involved in DNA replication leads to replication fork stalling and improper processing of nascent DNA fragments, resulting in chromosome aberrations, genomic instability and improper chromosome segregation and cell death Citation[21–23]. Disruption of proteins involved in DNA repair leads to an increase in unrepaired DNA damage. Ultimately unrepaired DNA damage contributes to genomic instability and results in chromosome aberrations.
Figure 1. Mathematical equations for hyperthermia survival curves. A typical hyperthermia survival curve is illustrated in the left panel. Two equations to describe the curve mathematically are shown on the right. While there is no there is no empirical difference between the equations in terms of fitting the data, there is a difference in the number of model parameters that are simple functions of absolute temperature (see text).
It is important to distinguish the effects, on proteins, of temperatures in the hyperthermic range, 40–47°C from the effects of thermal ablation temperatures (48–60°C) on proteins. Temperatures in the hyperthermic range cause proteins to be unfolded, exposing hydrophobic groups, which can then interact forming aggregates (). These heat effects on proteins are, in principle, reversible by the presence of molecular chaperones, such as HSP 70 Citation[24–25]. In the case of thermal ablation, severe protein denaturation is occurring and largely these protein changes are irreversible. The main evidence for protein unfolding aggregation comes from the studies by Jim Lepock using scanning calorimetry Citation[26–27] and the model system proteins Citation[28–29]. Using differential scanning calorimetry, it is possible to detect the endothermic reactions within a cell. The endothermic reactions are due to the denaturation or change of state in proteins in the cell. Peaks in the differential calorimetry curves, reflecting protein unfolding can be seen at temperatures in the 45–55°C range, 57–65°C temperature range, 65–75°C, 85°C and 100°C range. Sensitizers to heat shift the differential scanning calorimetry curves to lower temperatures, whereas thermal protectors shift these curves to higher temperatures Citation[30–31]. One interesting point that comes from studying these endothermic transitions is that a hyperthermia time/temperature combination that reduces survival by 10−2 can occur when ∼10% of the endothermic cellular transitions have occurred Citation[26]. Thus, the lower temperature transitions have a significant impact on cell survival.
Does hyperthermia cause DNA damage?
It has long been recognized that higher temperatures in the 50–60°C range induce DNA damage and denaturation Citation[32–34]. However, early work on DNA damage after hyperthermia in the 40–46°C range, showed that although heat does induce chromosome aberrations in S-phase cells Citation[22], Citation[34], DNA damage could not be detected by direct measurement Citation[36] or when detected could not be attributed to a direct effect on DNA Citation[37]. Thus the effects of hyperthermia on DNA in chromatin have been thought to be not due to direct heat-induced DNA damage, but secondary to heat effects on proteins involved in DNA replication, chromosome segregation and DNA repair Citation[22]. However, this idea has become somewhat controversial in the last year due to a paper reporting the formation of γH2AX foci as a result of hyperthermia and suggesting that these foci were markers for DNA double strand breaks Citation[38]. Since the formation of γ-H2AX foci is more sensitive to radiation dose than the classical DNA damage techniques Citation[39], it was conceivable that these techniques failed to detect DNA damage that was detected as γH2AX foci. To understand this issue it is important to realize that γH2AX is a modification of chromatin that occurs in the vicinity of a DNA double strand break. Therefore, γH2AX foci are, in fact, measuring a phosphorylation change in protein, which, in turn, is thought to reflect the presence of a DNA double strand break. A recent publication Citation[39] showed that heat shock (43°C for 30 or 60 min) induced γH2AX foci but not DNA double strand breaks detectable by pulse field gel electrophoresis. Interestingly, the γH2AX foci formation as the result of heat shock was dependent on the presence of the ATM molecule, whereas in cells lacking ATM, these foci did not occur. In contrast, following ionizing radiation, in which the formation of DNA double strand breaks is well established, γH2AX foci form independently of the presence of the ATM protein Citation[39]. Another striking difference between the γH2AX foci induced by hyperthermia and those induced by ionizing radiation is that those induced by hyperthermia do not support the finding of 53BP1, a molecule that is necessary for the assembly of other proteins required for repair of the double strand break Citation[39]. Interestingly, the γH2AX foci from both hyperthermia and ionizing radiation include the molecule, MDC1 so the conclusion of these studies is that heat induces foci containing γH2AX and MDC1 in the absence of DNA double strand breaks and these foci are inherently different from those which form in response to DNA double strand breaks. However, the exact nature of the differences between ionizing radiation-induced γH2AX foci and those induced by hyperthermia remains to be delineated. From these studies we conclude that hyperthermia does not directly induce DNA damage. However, the fact that heated S-phase cells develop chromosome aberrations suggests that DNA damage (resulting in chromosome aberrations 34,40) could arise secondary to the effects of heat on DNA replication.
Figure 2. Thermal effects on protein folding. Panel A is an illustration of a model for protein unfolding under hyperthermic conditions, in contrast to the conditions that apply for thermal ablation. As part of the evidence for this model, Panel B shows differential scanning calorimetry data. The peaks represent endothermic transitions. (See text for additional evidence.) Panel C shows the integral of all the endothermic transitions. It can be seen that more than 60% of the endothermic cellular transitions occur in the thermal ablation temperature range 60–70°C. In contrast, less than 10% of the transitions have occurred in the hyperthermic range even for temperatures that reduce survival by 10−2 in less than 15 min Citation[14].
![Figure 2. Thermal effects on protein folding. Panel A is an illustration of a model for protein unfolding under hyperthermic conditions, in contrast to the conditions that apply for thermal ablation. As part of the evidence for this model, Panel B shows differential scanning calorimetry data. The peaks represent endothermic transitions. (See text for additional evidence.) Panel C shows the integral of all the endothermic transitions. It can be seen that more than 60% of the endothermic cellular transitions occur in the thermal ablation temperature range 60–70°C. In contrast, less than 10% of the transitions have occurred in the hyperthermic range even for temperatures that reduce survival by 10−2 in less than 15 min Citation[14].](/cms/asset/9af52b65-6878-4e29-9559-c1548e5664f8/ihyt_a_276976_f0002_b.gif)
Figure 3. Two disparate heat effects on S-phase progression. Hyperthermia causes a greater delay in the completion of DNA synthesis (Panel B) than in the transition from a type I DNA replication pattern to a type II DNA replication pattern. This differential delay causes mis-timing of DNA replication within S-phase and means that early S and mid S DNA replication factories are synthesizing DNA at the same time indicating a loss of check point control. (From Citation[47], used with permission.)
![Figure 3. Two disparate heat effects on S-phase progression. Hyperthermia causes a greater delay in the completion of DNA synthesis (Panel B) than in the transition from a type I DNA replication pattern to a type II DNA replication pattern. This differential delay causes mis-timing of DNA replication within S-phase and means that early S and mid S DNA replication factories are synthesizing DNA at the same time indicating a loss of check point control. (From Citation[47], used with permission.)](/cms/asset/04d56973-2cbc-4676-89c2-834800a2b005/ihyt_a_276976_f0003_b.gif)
Effects of hyperthermia on DNA replication and cell cycle progression
It is recognized that heat has numerous affects on DNA replication. The pioneering work of Dewey et al. Citation[34] showed that S-phase cells are hypersensitive to heat induced killing and that this killing correlates with chromosome aberrations Citation[5] Citation[21–22], which, as noted above, are induced when cells are heated in S-phase. At clinically achievable temperatures ≤42.5°C the major component of cell killing will be the killing of S-phase cells Citation[40–42]. For example when cells are continuously heated at 41°C, the prominent observed effect is an S-phase block, after 24 h at 41.5°C, 80% of the cells occur in S-phase and cells will enter mitosis prior to the completion of DNA replication and this process will result in cell death by mitotic catastrophe Citation[43]. Interestingly, the hypersensitivity of S-phase cells to heat shock appears to be due to potentially lethal lesions, i.e. lesions that can be repaired unless they are fixed to lethal lesions before repair is complete. Specifically, if a cell in S-phase can be prevented from making DNA following heat shock for a long enough period of time (2–6 h), the thermal sensitivity of S-phase cells is essentially the same as a non-S-phase cell Citation[44]. Thus, the thermal damage responsible for S-phase hypersensitivity can be essentially completely reversed if it can be repaired. Conversely DNA replication appears to fix the potentially lethal lesions into lethal ones. In other words DNA replication in the presence of the potentially lethal lesion renders it lethal, while reversibly arresting DNA replication allows time for the lesion to be repaired. The time course for repair of this potential lethal damage is 2–6 h, depending on the severity of the damage. For example, after a 15 min, 45°C heat shock, the hypersensitivity of S-phase cells can be reversed by blocking DNA synthesis for 2 h Citation[44]. However, after a 30 min at 45°C heat shock, the hypersensitivity of S cells can only be reversed after 6 h of inhibition of DNA synthesis Citation[44]. Thus, the thermal sensitivity of S-phase cells is due to a condition that can be repaired. DNA synthesis is delayed long enough for the repair to be completed.
Figure 4. Effects on hyperthermia on DNA Supercoiling Ability. (a) illustrates a model of how hyperthermia induced MAR DNA binding can impact DNA supercoiling and the recognition of DNA damage. In the absence of the heat-induced MAR binding proteins single-strand DNA breaks will inhibit DNA supercoil rewinding, but when these proteins bind MAR DNA the effects of these breaks on DNA supercoiling are masked, see (b) which shows some of the supporting data in which DNA damage induced by ionizing radiation is masked in heated cells. The excess halo diameter is illustrated in (c), showing the approximate point at which the difference between control and irradiated samples were measured and added to estimate the difference between the curves. Note that in the samples from heated and irradiated cells the inhibition of DNA supercoil rewinding is reversed due to the masking of the DNA damage. Panel C reprinted from Citation[39].
![Figure 4. Effects on hyperthermia on DNA Supercoiling Ability. (a) illustrates a model of how hyperthermia induced MAR DNA binding can impact DNA supercoiling and the recognition of DNA damage. In the absence of the heat-induced MAR binding proteins single-strand DNA breaks will inhibit DNA supercoil rewinding, but when these proteins bind MAR DNA the effects of these breaks on DNA supercoiling are masked, see (b) which shows some of the supporting data in which DNA damage induced by ionizing radiation is masked in heated cells. The excess halo diameter is illustrated in (c), showing the approximate point at which the difference between control and irradiated samples were measured and added to estimate the difference between the curves. Note that in the samples from heated and irradiated cells the inhibition of DNA supercoil rewinding is reversed due to the masking of the DNA damage. Panel C reprinted from Citation[39].](/cms/asset/9981d6c2-8cbb-4c67-9e45-4fa0ece46707/ihyt_a_276976_f0004_b.gif)
The role of DNA replication fork stalling secondary to the enhanced protein binding in DNA replication complexes as a mechanism for the hypersensitivity of S-phase cells to hyperthermia
To gain insight into the fixing of heat-induced lethal lesions by DNA replication, we considered the effects of hyperthermia on DNA replication. Nuclear DNA replication is coordinated in time and space and associated with the nuclear matrix. Nuclear DNA is replicated in large protein comp-lexes, called replication factories. These replication factories have different organizations within the nucleus depending on whether the cell is in early, mid or late S-phase Citation[45–46]. For example, actively replicating DNA in early S-phase, DNA replication complexes can be detected (by pulse-labelling with BUdR and staining with a fluorescently labelled antibody to DNA containing BU) as small foci distributed throughout the nucleus, called a type I DNA replication pattern Citation[44], Citation[46]. In mid S-phase active replication complexes are detected in the nuclear periphery and observed in larger aggregates than the foci representative of early S-phase. The mid-S phase replication patterns are called a type II DNA replication pattern Citation[44], Citation[46]. In late S-phase the active replication complexes occur in even larger aggregates, called a type III DNA replication pattern Citation[44], Citation[46]. Hyperthermia can disrupt the timing of the transitions between the Type I, II, and III patterns Citation[45]. Specifically, if one compares the heat induced delay in transition from Type I to Type II DNA replication patterns, one sees a lack of dose dependent delay and a rather short delay of about 2 h, whereas in the case of overall DNA synthesis, this delay is heat-dose dependent and can take up to 12 h for a cell that has been heated at 45°C for 30 min to replicate half of its DNA Citation[47]. This result suggests that the cells were initiating DNA replication complexes scheduled to start in mid S-phase cells prior to the completion of replication by the factories that are scheduled to make DNA in early S. To verify this idea, a double pulse labelled technique was used, using iodo-deoxyuridine and chloro-deoxyuridine to distinguish the actively replicating complexes at two different times. In unheated cells, if the time between the labels is 4 h the labels are completely separated, showing that Type II DNA replication factories do not initiate DNA synthesis until almost all of the Type I replication factories have completed DNA replication Citation[47]. We were able to show that if the labels were separated by 6.5 h, the second label picked up both Type I and Type II DNA replication complexes, showing that Type II DNA replication factories had initiated DNA synthesis while most of the Type I replication factories were still replicating DNA Citation[47]. Thus, heat shock induces a disruption of the timing and coordination of DNA replication as cells progressed through S.
To gain insight into the heat induced lesion that is repairable in the absence of DNA replication, we considered the effects of hyperthermia on selected proteins involved in DNA replication. If one looks at the tightness of binding of DNA replication proteins within the factories and to the nuclear matrix, one can see that hyperthermia has induced a solubility change in the proteins involved in DNA replication Citation[47–48]. In other words, binding of these proteins is tighter, their associations are less fluid and presumably their ability to function is impaired. These solubility changes can be measured under different conditions of heat-dose responses and recovery times in the absence of DNA replication and in this particular study, cells were given various heat exposures and either allowed to continue to synthesize DNA immediately or held under conditions that arrest DNA synthesis for 4 h and then allowed to continue DNA synthesis. Alternatively, cells were given either a 15 min at 45°C or 30 min at 45°C heat shock and kept from continuing DNA synthesis for various time intervals. At the end of each heat exposure and DNA synthesis arrest period, cell survival and the binding of selected DNA replication proteins to the nuclear matrix were measured. Note that tighter binding to the nuclear matrix reflected the binding of proteins in DNA replication complexes. For proteins involved in DNA replication, binding to the nuclear matrix correlated with cell killing, whereas such binding of proteins not involved in DNA replication did not correlate with cell killing suggesting that the repairable, potentially lethal lesion is tighter binding of proteins throughout the DNA replication factories Citation[43], Citation[46]. Thus, it is likely that the protein binding causes an ineffective DNA replication complex, resulting in stalling of DNA replication forks, which can create DNA double strand breaks. It is known that DNA replication forks create double strand breaks and that homologous recombination is required to repair such breaks Citation[49]. So the question then becomes: in S-phase cells, is γH2AX foci higher in S phase cells as opposed to non-S phase cells? Recent studies Citation[37], Citation[39] have found that S phase cells have a higher number of γH2AX foci compared to G1 or G2 cells for the same heat exposure. Thus, it seems that γH2AX foci in S phase cells consist of a combination of heat-induced foci per se, and to foci formed as a result of DNA double strand breaks secondary to stalled DNA replication forks. This issue should be an attractive one for further research because the story is highly suggestive, but few of the details are available at present.
Interestingly, other effects of hyperthermia on cell cycle progression occur in addition to the effects of heating of S-phase. After 15 min at 45°C, when the cells are cultured at 37°C, there is a transient G2 block which lasts about 10–15 h after heat shock Citation[50]. Although few cells not in the S-phase are killed by this temperature, death appears to be related to failure to complete G2 in the cells heated by S-phase. After a 30 min at 45°C heat shock, again recovering at 37°C, there is a long lasting G2 and late S-phase block that persists for 14–35 h after the heat shock. Again cell death appears to be related to failure in exiting late G2 Citation[51].
Figure 5. Heat-induced mitochondrial damage and oxidative stress. (a) shows an overview of heat effects on mitochondria leading to a burst of reactive oxygen radicals (ROS). (b) shows novel functionalized indoles. (c) shows the effects of VJ112-OH in combination with 41°C hyperthermia. (d) shows that increasing the burden of ROS destabilizes proteins as measured by differential scanning calorimetry using a model system, in which the redox status of a C++ dependent ATPase had been altered so that the effects of redox status on the endothermic transitions characteristic of unfolding could be observed.
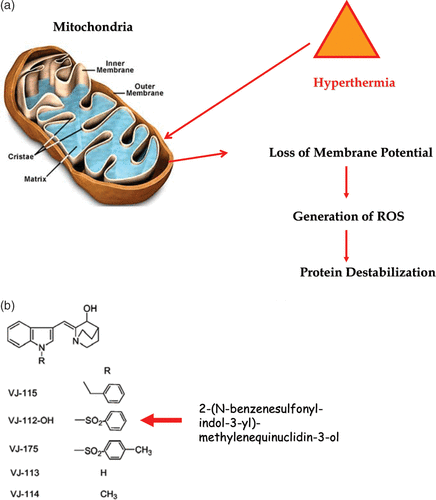
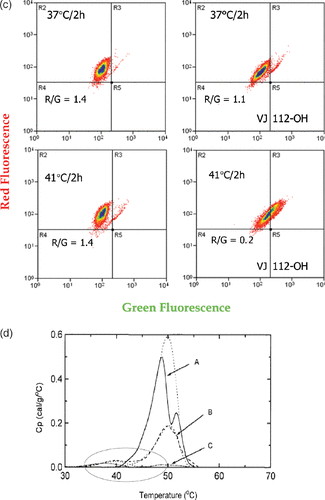
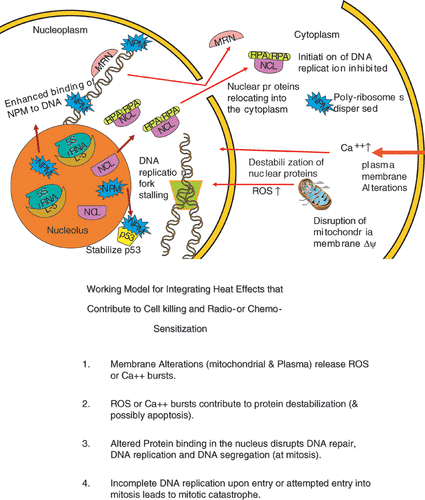
Figure 6. An illustration integrating the examples of hyperthermia effects discussed in the text. (a) is a simplified drawing of the effects described. (b) is a statement of an integrating model linking effects in the plasma membrane and the mitochondria to protein effects in the nucleus, cell killing and radio- or chemo- sensitization.
Effects on proteins associated with the nuclear matrix
It is known that heat induces a significant increase in proteins bound to the nuclear matrix Citation[52–56]. Citation[29], Citation[57] The nuclear matrix is defined as an organization structure within the nucleus that organizes the DNA into domains and supports most DNA related functions, such as transcription and replication Citation[17–20]. Aberrant protein binding to this structure has the potential to disrupt a large number of nuclear functions. Transcription and RNA processing, as well as DNA replication are known to be inhibited by heat shock Citation[58–59]. Citation[21]The approaches that have been used to detect the effects of hyperthermia on proteins associated with the nuclear matrix are alterations in protein solubility Citation[52–56], Citation[29], Citation[57] protein-DNA interactions Citation[60], flow cytometry Citation[61], the effects of heat shock on DNA supercoiling ability Citation[62–64] and differential scanning calorimetry Citation[14]. Lepock and Warters have shown that using differential scanning calorimetry on isolated nuclei and isolated nuclear matrix preparations that the nuclear matrix is among the more heat sensitive organelles showing significant endothermic changes as low as 40°C Citation[14]. If we look at the additional proteins that bind nuclear matrix associated DNA after heat shock, these proteins appear to mask DNA strand breaks delaying repair and alter DNA supercoiling changes that may be a critical trigger for DNA repair Citation[65] and are known to be critical for DNA replication and transcription Citation[66].
Changes in the associations of nuclear proteins and protein DNA interactions and proteins binding to the nuclear matrix do not occur in isolation from other heat effects within the cell. It has long been recognized that heat has effects on plasma membrane protein distribution and alters the permeability of plasma membranes resulting in a calcium spike Citation[67–70]. There are also significant disruptions of mitochondrial membrane potential resulting in the change in the redox status of cells Citation[71–72]. It has been shown by Mike Freeman that this change in redox status can sensitize cells to hyperthermia Citation[73–74]. This work further showed that the mitochondrial membrane depolarization and the resulting burst of reactive oxygen species (ROS) i.e. oxygen radicals alter protein stability rendering proteins sensitive to heat-induced protein unfolding. The altered protein stability can destabilize proteins within the nucleus disrupting DNA repair, DNA replication and DNA segregation. DNA double strand break formation secondary to heat-induced DNA replication fork stalling can lead to lethal lesions. There is also evidence that inhibition of DNA replication coupled with heat induced defects in checkpoint signalling leads to incomplete DNA replication upon entry into mitosis, which leads to mitotic catastrophe. This is a simple integrated model of the killing of S-phase cells by heat that explains the correlation between cell killing and chromosome aberrations Citation[13]. Many aspects of this model have been verified over the years Citation[22], Citation[74]. Further support from the model has been derived from recently developed thermal sensitizing compounds based on indomethacin analogs Citation[76]. These compounds have been shown to sensitize the mitochondrial membrane to heat induced disruption and at the same time decrease the temperatures for thermal destabilization of proteins, alter the binding of DNA repair proteins and reduce the temperature required to cause a delay in the assembly of DNA repair complexes (Laszlo and Freeman, unpublished). The mechanisms of these compounds appear to be consistent with the idea that heat effects on membranes, mitochondria and nuclear proteins are interrelated. The above considerations do not explain how heat alters viability in G1, and G2 cells, which remains to be addressed in detail.
Redistribution of proteins from the nucleus
In contrast to inducing tighter binding of a large number of proteins throughout the nucleus, hyperthermia can also loosen the binding of certain nuclear proteins and drive them out of the nucleus. Two examples of this effect are the DNA repair protein, Mre11, and the nucleolar protein, nucleolin (NCL). Temperatures of 41°C or higher will drive Mre11 out of the nucleus and lead to a subsequent sensitization to ionizing radiation Citation[77–80]. This effect could be related to heat dissociating Mre11 from its functional complex with other DNA repair proteins Citation[81]. As evidence for this possibility, heat induces an association between Mre11 and HSP70 Citation[81]. In the case of NCL, heat induces dissociation between NCL and its functional partner nucleophosmin (NPM) and an association with the DNA replication protein, RPA Citation[82]. This complex will then be exported from the nucleus. It has been suggested that this is a mechanism to prevent DNA replication, as RPA is needed for replication Citation[82]. As discussed above, DNA replication can result in the fixing of potential lethal lesions to lethal lesions and it is speculated that the RPA/NCL binding mechanism may prevent heated cells from entering S phase, where such potential lethal lesions could be fixed. These considerations indicate that heat can disrupt the functions of large protein complexes by either or both tightening the some proteins throughout the complex or by decreasing the association of a critical member of the complex. These individual circumstances then become important areas for hyperthermic research.
Summary
The challenge of understanding of how hyperthermia induces cell killing, inhibits DNA repair and sensitizes cells to chemotherapeutic agents represents an ongoing challenge, because of the numerous protein changes induced by hyperthermia and the fact that it is likely that a small fraction of these changes will be relevant to the endpoint of concern to the investigator. However, much progress has been made in the understanding of hyperthermia effects on cellular processes. Thus, research on the mechanisms of hyperthermic effects becomes more sophisticated as better understanding of cellular processes of interest to the investigator become available. A better understanding of how hyperthermia alters cellular processes relevant to therapeutic endpoints will facilitate a more effective employment of hyperthermia as a therapeutic adjuvant.
References
- Coffey DS, Getzenberg RH, DeWeese TL. Hyperthermic biology and cancer therapies: A hypothesis for the ‘Lance Armstrong effect’. JAMA 2006; 296: 445–48
- Sapareto SA, Hopwood LE, Dewey WC, Raju MR, Gray JW. Effects of hyperthermia on survival and progression of Chinese hamster ovary cells. Cancer Res 1978; 38: 393–400
- Raaphorst GP, Freean ML, Dewey WC. Radiosensitivity and recovery from radiation damage in cultured CHO cells exposed to hyperthermia at 42.5 or 45.5°C. Radiat Res 1979; 79: 390–402
- Henle KJ, Roti Roti JL. Response of cultured mammalian cells to hyperthermia. Hyperthermia and Oncology, M Urano, E Douple. VSP, Thermal effect on cells and tissues, The Netherlands 1988; 1: 57–82
- Dewey WC, Hopwood LE, Sapareto SA, Gerweck LE. Cellular responses to combinations of hyperthermia and radiation. Radiology 1977; 123: 463–479
- Henle KJ, Dethlefsen LA. Time-temperature relationships for heat-induced killing of mammalian cells. Ann NY Acad Sci 1980; 335: 234–253
- Roti Roti JL, Henle KJ. Comparison of two mathematical models for describing heat-induced cell killing. Radiat Res 1980; 81: 374–383
- Mackey MA, Roti Roti JL. A model of heat-induced clonogenic cell death. J Theor Biol 1992; 156: 133–146
- Harmon BV, Corder AM, Collins RJ, Gobe GC, Allen J, Allan DJ, Kerr JF. Cell death induced in a murine mastocytoma by 42–47°C heating in vitro: evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol 1990; 58: 845–858
- Coss RA, Dewey WC, Bamburg JR. Effects of hyperthermia 41.5°C on Chinese hamster ovary cells analyzed in mitosis. Cancer Res 1979; 39: 1911–1918
- Mackey MA, Ianzini F. Enhancement of radiation-induced mitotic catastrophe by moderate hyperthermia. Int J Radiat Biol 2000; 76: 273–280
- VanderWaal R, Malyapa RS, Higashikubo R, Roti Roti JL. A comparison of modes and kinetics of cell death in L5178Y and HeLa cells following hyperthermia. Radiat Res 1997; 148: 455–462
- Roti Roti JL, Laszlo A. The effects of hyperthermia on cellular macromolecules. Hyperthermia and Oncology, M Urano, E Douple. VSP, Thermal effect on cells and tissues, The Netherlands 1988; 1: 13–56
- Lepock JR. Role of nuclear protein denaturation and aggregation in the thermal radiosensitization. Int J Hyperthermia 2004; 20: 115–130
- Pienta KJ, Getzenberg RH, Coffey DS. Cell structure and DNA organization. Crit Rev Eukaryot Gene Expr 1991; 1: 355–385
- Berezney R, Coffey DS. Identification of a nuclear protein matrix. Biochem Biophys Res Commun 1974; 60: 1410–1417
- Berezney R, Mortillaro MJ, Ma H, Wei X, Samarabandu J. The nuclear matrix: A structural milieu for nuclear genomic function. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 1–63
- Nickerson JA, Blencowe BJ, Penman S. The architectural organization of nuclear metabolism. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 67–124
- Jackson DA, Cook PR. The structural basis of nuclear function. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 125–160
- Agutter PS. Intracellular structure and nucleocytoplasmic transport. Nuclear matrix: Structural and functional organization, R Berezney, K Jeon. Academic Press, California 1995; 183–224
- Wong RSL, Kapp LN, Dewey WC. DNA for displacement rate measurements in heated Chinese hamster ovary cells. Biochim Biophys Acta 1989; 1007: 224–227
- Wong RSL, Kapp LN, Krishnaswamy G, Dewey WC. Critical steps for induction of chromosomal aberrations in CHO cells heated in S phase. Radiat Res 1003; 133: 52–59
- Wong RSL, Thompson LL, Dewey WC. Recovery from effects of heat on DNA synthesis in Chinese hamster ovary cells. Radiat Res 1988; 114: 125–137
- Stege GJ, Li L, Kampinga HH, Konings AW, Li GC. Importance of the ATP binding domain and nucleolar localization domain of HSP72 in the protection of nuclear proteins against heat-induced aggregation. Exp Cell Res 1994; 21: 279–284
- Kampinga, HH, Brunsting JF, Stege GJ, Konings AW, Landry J. Cells overexpressing Hsp27 show accelerated recovery from heat-induced nuclear protein aggregation. Biochem Biophys Res Commun 1994; 204: 1170–1177
- Lepock JR, Frey HE, Rodahl AM, Kruuv J. Thermal analysis of CHL V79 cells using differential scanning calorimetry: Implications for hyperthermia cell killing and the heat shock response. J Cell Physiol 1988; 137: 14–24
- Lepock JR, Frey HE, Ritchie KP. Protein denaturation in intact hepatocytes and isolated cellular organelles during heat shock. J Cell Biol 1993; 122: 1267–1276
- Michels AA, Nguyen VT, Konings AWT, Kampinga HH, Bensaude O. Thermostability of a nuclear-targeted luciferase expressed in mammalian cells: Destabilizing influence of the intranuclear microenvironment. Europ J Biochem 1995; 234: 382–389
- Lepock JR, Frey HE, Heynen MP, Nishio J, Waters B, Ritchie KP, Kruuv J. Increased thermostability of thermotolerant CHL V79 cells as determined by differential scanning calorimetry. J Cell Physiol 1990; 142: 628–634
- Borrelli MJ, Lepock JR, Frey HE, Lee YJ, Corry PM. Excess protein in nuclei isolated from heat-shocked cells results from a reduced extractability of nuclear proteins. J Cell Physiol 1996; 167: 369–379
- Klump H. Thermodynamic values of the helix-coil transition of DNA in the presence of quaternary ammonium salt. Biochim Biophys Acta 1977; 475: 605–610
- Lewis PN. A thermal denaturation study of chromatin and nuclease-produced chromatin fragments. Can J Biochem 1977; 55: 736–746
- Defer N, Kitzis A, Kruh J, Brahms S, Brahms J. Effect of non-histone proteins on thermal transition of chromatin and of DNA. Nucleic Acids Res 1977; 4: 2293–2306
- Dewey WC, Westra A, Miller HH, Nagasawa H. Heat-induced lethality and chromosomal damage in synchronized Chinese hamster cells treated with 5-bromodeoxyuridine. Int J Radiat Biol 1971; 20: 505–520
- Warters RL, Brizgys LM, Axtell-Bartlett J. DNA damage production in CHO cells at elevated temperatures. J Cell Physiol 1985; 123: 481–486
- Jorritsma JB, Konings AW. The occurrence of DNA strand breaks after hyperthermic treatments of mammalian cells with and without radiation. Radiat Res 1940; 98: 198–208
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, Mori E, Yamakawa N, Yasumoto J, Yuki K, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004; 64: 8839–8845
- Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol 2004; 75: 355–373
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, Gupta A, Rief N, Horikoshi N, Baskaran R, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 6: 3010–3017
- Mackey MA, Anolik SL, Roti Roti JL. Changes in heat and radiation sensitivity during long duration, moderate hyperthermia in HeLa S3 cells. Int J Radiat Biol Oncol Phys 1992; 24: 543–550
- Mackey MA, Roti Roti JL. A model of heat-induced clonogenic cell death. J Theor Biol 1992; 156: 133–146
- Mackey MA, Dewey WC. Time-temperature analyses of cell killing of synchronous G1 and S phase Chinese hamster cells in vitro. Radiat Res 1988; 113: 318–333
- Mackey MA, Zhang XF, Hunt C, Sullivan S, Blum J, Laszlo A, Roti Roti JL. Uncoupling of M-phase kinase activation from the completion of S phase by heat shock. Cancer Res 1996; 56: 1770–1774
- VanderWaal RP, Griffith CL, Wright WD, Higashikubo R, Borrelli MJ, Roti Roti JL. Delaying S-phase progression rescues cells from heat-induced S-phase hypertoxicity. J Cell Physiol 2001; 187: 236–243
- Berezney R. Visualizing DNA replication sites in the cell nucleus. Sem Cell Biol 1991; 2: 103–115
- Wei X, Samarabandu J, Devdhar RS, Siegel AJ, Acharya R, Berezney R. Segregation of transcription and replication sites into higher order domains. Science 1998; 281: 1502–1506
- vanderWaal RP, Wright WD, Roti Roti JL. The effects of heat-shock on nuclear-matrix-associated DNA-replication complexes. Crit Rev Eukaryotic Gene Expression 1999; 9: 363–371
- vanderWaal RP, Higashikubo R, Xu M, Roti Roti JL. Cytometric methods to analyze thermal effects. Methods in Cell Biology 2001; 64: 269–286
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai M, Shinohara A, Takeda S. Homologous recombination and non-homologous en-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17: 5497–5508
- Roti Roti JL, Uygur N, Higashikubo R. Nuclear protein following heat shock: Protein removal kinetics and cell cycle rearrangements. Radiat Res 1986; 107: 250–261
- Iliakis GE, Pantelias GE. Effects of hyperthermia on chromatin condensation and nucleoli disintegration as visualized by induction of premature chromosome condensation in interphase mammalian cells. Cancer Res 1989; 49: 1254–1260
- Roti Roti JL, Winward RT. The effects of hyperthermia on the protein to DNA ratio of isolated HeLa chromatin. Radiat Res 1978; 74: 159–169
- Tomasovic SP, Turner GN, Dewey WC. Effects of hyperthermia on non-histone proteins isolated with DNA. Radiat Res 1978; 73: 535–552
- Blair OC, Winward RT, Roti Roti JL. The effect of hyperthermia on the protein content of HeLa nuclei: A flow cytometric analysis. Radiat Res 1979; 78: 474–484
- Kampinga HH, Jorritsma JB, Konings AW. Heat-induced alterations in DNA polymerase activity of HeLa cells and of isolated nuclei. Relation to cell survival. Int J Radiat Biol Relat Stud Phys Chem Med 1985; 47: 29–40
- Chu GL, Ross G, Wong WSL, Warters R, Dewey WC. Content of nonhistone protein in nuclei after hyperthermic treatment. J Cell Phys 1993; 154: 217–221
- Roti Roti JL, Higashikubo R, Mace M. Protein cross-migration during isolation of nuclei from mixtures of heated and unheated HeLa cells. Radiat Res 1984; 98: 107–114
- Sakkers RJ, Brunsting JF, Filon AR, Kampinga HH, Konings AW, Mullenders LH. Altered association of transcriptionally active DNA with the nuclear-matrix after heat shock. Int J Radiat Biol 1999; 75: 875–883
- Sadis S, Hickey E, Weber LA. Effect of heat shock on RNA metabolism in HeLa cells. J Cell Physiol 1988; 135: 377–386
- Vanderwaal RP, Roti Roti JL. Heat induced ‘masking’ of redox sensitive component(s) of the DNA-nuclear matrix anchoring complex. Int J Hyperthermia 2004; 20: 234–239
- Wright WD, Higashikubo R, Roti Roti JL. Flow cytometric studies of the nuclear matrix. Cytometry 1989; 10: 303–311
- Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (> or = 43°C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 2004; 20: 131–139
- Roti Roti JL, Painter RB. Effects of hyperthermia on the sedimentation of nucleoids from HeLa cells in sucrose gradients. Radiat Res 1981; 89: 166–175
- Roti Roti JL, Wright WD. Visualization of DNA loops in nucleoids from HeLa cells: Assays for DNA damage and repair. Cytometry 1987; 8: 461–467
- Bakkenist CJ, Kasten MB. DNA damage activates ATM through intermolecular autophosphorylation and dimmer dissociation. Nature 2003; 421: 499–506
- Vogelstein B, Pardoll DM, Coffey BS. Supercoiled loops and eukaryotic DNA replication. Cell 1980; 22: 799–805
- Calderwood SK, Stevenson MA, Hahn GM. Effects of heat on cell calcium and inositol lipid metabolism. Radiat Res 1988; 113: 414–425
- Vidar CA, Dewey WC. Evaluation of a role for intracellular Na + , K + , Ca2 + and Mg2_ in hyperthermic cell killing. Radiat Res 1986; 105: 187–200
- Mikkelsen RB, Stedman T. Cytotoxic hyperthermia and Ca2 + homeostasis: The effect of heat on Ca2 + uptake by nonmitochondrial intracellular Ca2 + stores. Radiat Res 1990; 123: 82–86
- Stege GJ, Wisrenga PK, Kampinga HH, Konings AW. Hyperthermia, intracellular free calcium and calcium ionophores. Int J Radiat Biol 1993; 64: 459–468
- Senisterra GA, Huntley SA, Escaravage M, Sekhar KR, Freeman ML, Borrelli M, Lepock JR. Destabilization of the Ca2 + -ATPase of sarcoplasmic reticulum by thiol-specific, heat shock inducers results in thermal denaturation at 37°C. Biochemistry 1997; 36: 11002–11011
- Dressler C, Beuthan J, Mueller G, Zabarylo U, Minet O. Fluorescence imaging of heat-stress induced mitochondrial long-term depolarization in breast cancer cells. J Fluoresc 2006; 16: 689–695
- Da-Yong Y, Matsuya Y, Zhao Q-L, Ahmed K, Wei Z-L, Nemoto H, Kondo T. Enhancement of hyperthermia-induced apoptosis by a new synthesized class of furan-fused tetracyclic compounds. Apoptosis 2007; 12: 1523–1532
- Nijhuis EH, Poot AA, Feijen J, Vermes I. Induction of apoptosis by heat and gamma-radiation in a human lymphoid cell line: Role of mitochondrial changes and caspase activation. Int J Hyperthermia 2006; 22: 687–698
- Dewey WC, Li X, Wong RSL. Cell killing, chromosomal aberrations and division delay as thermal sensitivity is modified during the cell cycle. Radiat Res 1990; 122: 268–274
- Sekhar KR, Sonar VN, Muthusamy V, Sasi S, Laszlo A, Sawani J, Horikoshi N, Higashikubo R, Bristow RG, Borrelli MJ, Crooks PA, Lepock JR, Roti Roti JL, Freeman ML. Novel chemical enhancers of heat shock increase thermal radiosensitization through a mitotic catastrophe pathway. Cancer Res 2007; 67: 695–701
- Zhu WG, Seno J, Beck BD, Dynlacht JR. Translocation of Mre11 from the nucleus to the cytoplasm as a mechanism of radiosensitization by heat. Radiat Res 2001; 156: 92–102
- Xu M, Myerson RJ, Straube WL, Moros EG, LaGroye I, Wang LL, Lee JT, Roti Roti JL. Radiosensitization of heat resistant human tumor cells by one hour at 41.1°C and its effect on DNA repair. Int J Hyperthermia 2002; 18: 385–403
- Xu M, Myerson RJ, Hunt C, Kumar S, Moros EG, Straube WL, Roti Roti JL. Transfection of human tumor cells with Mre11 siRNA increases radiation sensitivity and reduces heat induced radiosensitization. Int J Hyperthermia 2004; 20: 157–162
- Seno JD, Dynlacht JR. Intracellular redistribution and modification of proteins of the Mre11/Rad50/Nbs1 DNA repair complex following irradiation and heat shock. J Cell Physiol 2004; 199: 157–170
- Dynlacht JR, Xu M, Pandita RK, Roti Roti JL. Effects of heat shock on the Mre11/Rad50/Nbs1 complex in irradiated or unirradiated cells. Int J Hyperthermia 2004; 20: 144–156
- Wang Y, Guan J, Wang H, Wang Y, Leeper D, Illiakis G. Regulation of DNA replication after heat shock by replication protein A-nucleolin interactions. J Biol Chem 2001; 267: 20579–20588