Abstract
Heat shock is one of the most effective radiosensitizers known. As a result, combination of heat with ionizing radiation (IR) is considered a promising strategy in the management of human cancer. The mechanism of heat radiosensitization has been the subject of extensive work but a unifying mechanistic model is presently lacking. To understand the cause of excessive death in irradiated cells after heat exposure, it is necessary to characterize the lesion(s) underlying the effect and to determine which of the pathways processing this lesion are affected by heat. Since DNA double strand breaks (DSBs) are the main cause for IR-induced cell death, inhibition of DSB processing has long been considered a major candidate for heat radiosensitization. However, effective radiosensitization of mutants with defects in homologous recombination repair (HRR), or in DNA-PK dependent non-homologous end joining (D-NHEJ), the two primary pathways of DSB repair, has led to the formulation of models excluding DSBs as a cause for this phenomenon and attributing heat radiosensitization to inhibition of base damage processing. Since direct evidence for a major role of base damage in heat radiosensitization, or in IR-induced killing for that matter, is scarce and new insights in DSB repair allow alternative interpretations of existing data with repair mutants, we attempt here a re-evaluation of the role of DSBs and their repair in heat radiosensitization. First, we reanalyse data obtained with various DSB repair mutants on first principles and in the light of the recent recognition that alternative pathways of NHEJ, operating as backup (B-NHEJ), substantially contribute to DSB repair and thus probably also to heat radiosensitization. Second, we review aspects of combined action of heat and radiation, such as modulation in the cell-cycle-dependent variation in radiosensitivity to killing, as well as heat radiosensitization as a function of LET, and examine whether the observed effects are compatible with DSB repair inhibition. We conclude with a model reclaiming a central role for DSBs in heat radiosensitization.
Introduction
The extensive use of ionizing radiation (IR) in the management of human cancer has generated wide interest in the development of radiosensitizers. The rationale behind this effort lies in the recognition that selective potentiation in the killing of tumor cells will allow the development of improved treatment strategies. Heat shock is one of the most effective radiosensitizers known and the development of hyperthermia as a cancer treatment modality generates unique opportunities and interest in combining heat with IR. Indeed, evaluation of the effects of heat on radiation response has been one of the most active research areas in the field for several decades. The goal of these efforts is an improved understanding of the mechanisms by which heat potentiates radiation-induced cell killing. Since heat affects all aspects of cellular metabolism, including all DNA repair pathways studied to date, the key task is to identify those mainly contributing to heat radiosensitization - defined as enhancement in radiation-induced lethality.
For the mechanistic elucidation of this phenomenon, it is important to understand how heat potentiates cell killing. An obvious hypothesis is that heat radiosensitizes by inhibiting the processing of highly lethal radiation-induced DNA lesions. Among the lesions induced by IR the DNA double strand break is thought to have the highest probability of causing cell death. This potential derives from the lack of an intact DNA strand to be used as template for repair, and from the precarious discontinuity it generates in the chromosome. Indeed, the formation of chromosome aberrations, most likely resulting from error-prone repair of DSBs, has been shown to nearly perfectly correlate with IR-induced cell killing, with one visible chromosomal aberration being equivalent to a lethal event Citation[1] (). Notably, this correlation remains unchanged throughout the cell cycle in cells exposed to heat (). The fact that identical types of aberrations are scored in cells exposed to radiation alone, as well as in cells exposed to heat plus radiation Citation[1] (), provides perhaps the strongest direct support for the notion that heat impairs the processing of IR lesions with high potential for chromosome aberration formation, such as the DSBs.
Figure 1. Correlation between chromosome aberration formation and cell killing in cells exposed to radiation alone (X), or to heat plus radiation (DX), in different phases of the cell cycle as indicated. Heat exposure (45.5°C) time was adjusted to give 40% survival in all cell populations tested. The results are from Dewey et al. Citation[1]. (A) Survival curves and the corresponding chromosome aberration induction curves (deletions plus exchanges) for synchronous CHO cells treated at Mitosis, as well as the G1- and S-phase of the cell cycle. The data show a good correlation between hyperthermic radiosensitization as measured by cell lethality and hyperthermic radiosensitization as measured by chromosomal aberrations. This provides indirect evidence that heat sensitizes cells to radiation by impairing the repair of lesions that have the potential of causing chromosomal aberrations. On the basis of their properties, DSBs are likely candidates for this effect. Note the enhanced radiosensitization of S-phase cells as compared to G1 cells, as well as the lack of radiosensitization in M-phase cells. The relevance of the observations with M cells is discussed later in the text. (B) The data of panel (A) re-plotted for survival as a function of chromosome aberrations per cell. Note that at a survival of 37%, which corresponds to one lethal event per cell, nearly one chromosome aberration is also recorded per cell.
![Figure 1. Correlation between chromosome aberration formation and cell killing in cells exposed to radiation alone (X), or to heat plus radiation (DX), in different phases of the cell cycle as indicated. Heat exposure (45.5°C) time was adjusted to give 40% survival in all cell populations tested. The results are from Dewey et al. Citation[1]. (A) Survival curves and the corresponding chromosome aberration induction curves (deletions plus exchanges) for synchronous CHO cells treated at Mitosis, as well as the G1- and S-phase of the cell cycle. The data show a good correlation between hyperthermic radiosensitization as measured by cell lethality and hyperthermic radiosensitization as measured by chromosomal aberrations. This provides indirect evidence that heat sensitizes cells to radiation by impairing the repair of lesions that have the potential of causing chromosomal aberrations. On the basis of their properties, DSBs are likely candidates for this effect. Note the enhanced radiosensitization of S-phase cells as compared to G1 cells, as well as the lack of radiosensitization in M-phase cells. The relevance of the observations with M cells is discussed later in the text. (B) The data of panel (A) re-plotted for survival as a function of chromosome aberrations per cell. Note that at a survival of 37%, which corresponds to one lethal event per cell, nearly one chromosome aberration is also recorded per cell.](/cms/asset/e73b8479-cea0-4362-a8f9-e4779a6da8fb/ihyt_a_278504_f0001_b.gif)
Despite their documented high killing potential, IR-induced DSBs are efficiently recognized and removed by the cell. Two conceptually distinct mechanisms repair DSBs in higher eukaryotes (): Homologous recombination repair (HRR) and non-homologous end joining (NHEJ). HRR Citation[2–8] requires extensive homology, normally found in the sister chromatid available during S and G2, and utilizes the products of the Rad52 epistasis group of genes. The function of HRR in mammalian cells is well documented by assays using model substrates and site-specific induction of DSBs Citation[9]. For randomly induced DSBs, such as those generated after exposure to ionizing radiation, the contribution of HRR is indirectly documented by the decreased radiosensitivity to killing in late S and G2 phase, as well as by the increased chromosome aberration formation in mutants deficient in HRR factors (reviewed in Citation[10], Citation[11]). However, the contribution of HRR to the actual removal of IR-induced DSBs remains less well defined and HRR mutants display no marked defects in DSB repair when analysed using physical methods of detection Citation[10], Citation[11]. We return to this point later in this review.
Figure 2. Outline of the main pathways of DNA DSB repair. In addition to HRR utilizing genes of the RAD52 epistasis group, cells of higher eukaryotes also employ NHEJ. There are two pathways of NHEJ: the classical pathway utilizes Ku and DNA-PKcs and carries out ligation using DNA Ligase IV (D-NHEJ). Other known components of this pathway include factors involved in end-processing (Tdp1, PNKP, Artemis) and DNA polymerization (Pol μ, λ). In addition to D-NHEJ, cells are capable of mediating end joining via a pathway operating as a backup (B-NHEJ). There is evidence that this pathway utilizes the DNA Ligase III/XRCC1/PARP-1 module.
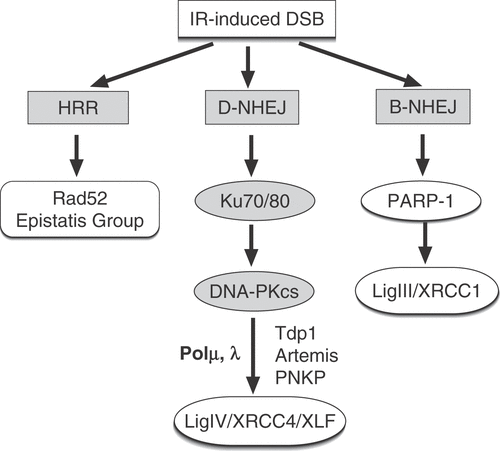
NHEJ requires no template and mediates end ligation after processing of the broken DNA termini Citation[4], Citation[12], Citation[13]. Central to NHEJ are the DNA-PK and DNA Ligase IV/XRCC4/XLF complexes, as well as PNKP, Tdp1 and Artemis and the DNA polymerases μ or λ (). This pathway is thought to operate throughout the cell cycle and to mediate impressively fast removal of DSBs Citation[14], Citation[15], albeit without ensuring restoration of the original sequence. For reasons that will become clear below, we proposed to refer to this pathway as D-NHEJ to indicate its dependence on DNA-PK and to distinguish it from similar pathways of DSB repair Citation[16], Citation[17].
To examine the contribution of different repair pathways to heat radiosensitization, mutants with defects either in D-NHEJ or HRR were studied. The initial rationale was that if the defective repair pathway were the only contributor to heat radiosensitization, the mutant will not be radiosensitized by heat – or it will only be radiosensitized to a limited extent. If, on the other hand, the mutant were defective in a repair pathway not contributing to heat radiosensitization, it will be radiosensitized to an extent similar to wild type cells through inhibition of the remaining repair activity.
The first report describing experiments conceived along these lines was published by our group using the radiosensitive mutant xrs-5, long before details regarding NHEJ and HRR were available in higher eukaryotes and the xrs-5 mutation pinpointed to the Ku80 gene Citation[18]. When this radiosensitive mutant was tested in the plateau phase of growth, heat radiosensitization was substantially reduced compared to its repair proficient counterpart, CHO (). This result, together with the observation that DNA repair processes were efficiently inhibited by heat in this cell line Citation[19], led to the hypothesis that the target lesion for heat radiosensitization is a subset of DSBs, and that the product of the xrs-5 gene (Ku80) contributes to a repair pathway (D-NHEJ) involved in heat radiosensitization Citation[18]. Specifically, it was proposed that DNA DSB proficiency is a prerequisite for heat radiosensitization.
Figure 3. The contribution of HRR and D-NHEJ to heat radiosensitization. (A) Survival curves of plateau-phase CHO cells irradiated and tested immediately after exposure to heat for the indicated times and temperatures. Note the significant heat radiosensitization achieved Citation[18]. (B) Similar data for the Ku80 deficient mutant xrs-5. Note the markedly reduced heat radiosensitization observed Citation[18]. (C) shows the effect of heat shock (45.5°C, 10 min) on the LIG4 deficient 180BRM, as well as in the repair-proficient MRC5SV1 tested during the exponential phase of growth. Note the extensive radiosensitization observed in the LIG4 deficient cells (see text for further discussion) (results obtained from Citation[25]). (D) shows the effect of heat exposure (43°C, 30 min) of the XRCC2 deficient mutant irs-1 and its wild type counterpart V79. Similar heat radiosensitization is observed in the parental cell line and in the HRR deficient mutant (see text for further discussion) (results obtained from Citation[25].
![Figure 3. The contribution of HRR and D-NHEJ to heat radiosensitization. (A) Survival curves of plateau-phase CHO cells irradiated and tested immediately after exposure to heat for the indicated times and temperatures. Note the significant heat radiosensitization achieved Citation[18]. (B) Similar data for the Ku80 deficient mutant xrs-5. Note the markedly reduced heat radiosensitization observed Citation[18]. (C) shows the effect of heat shock (45.5°C, 10 min) on the LIG4 deficient 180BRM, as well as in the repair-proficient MRC5SV1 tested during the exponential phase of growth. Note the extensive radiosensitization observed in the LIG4 deficient cells (see text for further discussion) (results obtained from Citation[25]). (D) shows the effect of heat exposure (43°C, 30 min) of the XRCC2 deficient mutant irs-1 and its wild type counterpart V79. Similar heat radiosensitization is observed in the parental cell line and in the HRR deficient mutant (see text for further discussion) (results obtained from Citation[25].](/cms/asset/2bf76481-0aef-4397-b2dc-09abb5a26d76/ihyt_a_278504_f0003_b.gif)
The lack of radiosensitization observed in xrs-5 cells, which pinpointed a lesion and a process for heat radiosensitization, could not be directly reproduced in a number of cell lines with defects in various components of D-NHEJ. Thus, embryonic fibroblasts from Ku80−/− mice showed levels of radiosensitization similar to those of wild-type cells Citation[20], Citation[21], Citation[22], and similar results were obtained with Ku70−/− DT40 cells Citation[20], Citation[23]. Also CHO cells defective in DNA-PKcs were found to be heat radiosensitized Citation[22], Citation[24] and similar results were again obtained with a DNA-PKcs−/− DT40 mutant Citation[23]. Even a human cell line with a hypomorphic mutation in LIG4 showed extensive radiosensitization Citation[21] (see ).
Testing of cells lines impaired in HRR gave similar results. Thus, cells with defects in XRCC3, a component of HRR, showed levels of radiosensitization similar to those of wild type cells Citation[20], as did also a V79 mutant with a defect in another component of HRR, XRCC2 Citation[25] (see also ). Also a Rad54−/− DT40 mutant showed marked radiosensitization Citation[20], Citation[23], as did a Rad54−/−/Ku70−/− double DT40 mutant Citation[20], Citation[23].
These results were invariably interpreted as evidence that inhibition of D-NHEJ or HRR is not a component of the mechanism of heat radiosensitization - a conclusion automatically ruling out DSBs as the lesions relevant for heat radiosensitization, if their repair can only proceed through these two pathways. On the basis of this conclusion and by applying deductive reasoning, arguments were developed that base damage plays a central role in heat radiosensitization Citation[25], Citation[26].
The above far reaching conclusion is based on several assumptions some of which are not explicitly spelled out or critically analysed in the light of experimental evidence. Furthermore, new insights into the mechanisms of DSB repair point to the operation of repair pathways in addition to D-NHEJ and HRR. These additional repair pathways allow in their own right alternative interpretations of heat radiosensitization results in radiosensitive DSB repair deficient mutants. For all these reasons, but also because base damage is not thought to play the central role in IR lethality that the chromosomal aberration results shown in would require, we attempt here a re-evaluation of the published evidence and develop counter-arguments that reclaim a central role for DSBs in heat radiosensitization.
Heat shock inhibits all pathways of DSB repair
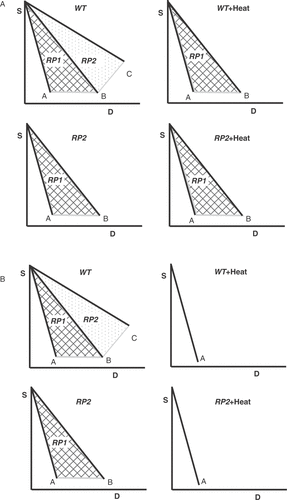
For an in-depth analysis of the role of DSBs as the target lesion for heat radiosensitization, it is important to discuss certain assumptions implicit in the interpretation of the results presented above. The conclusion that the pathway under investigation is not contributing to heat radiosensitization if the corresponding mutants can be radiosensitized requires that there is no other pathway available to repair DSBs, or that other available and still operational pathways are not heat sensitive. This is graphically illustrated in under the assumption that DSBs are the relevant lesions for heat radiosensitization and that two pathways are involved in their repair. If DSBs are repaired by a heat sensitive and a heat resistant pathway, as illustrated in panel A, no radiosensitization will be expected in a mutant with a completely compromised heat sensitive pathway RP2. This scenario is implicit in the interpretation of results reviewed above with cells deficient in either D-NHEJ or HRR, but as we will see below it is unlikely that the required conditions are satisfied.
Figure 4. Theoretical analysis of heat radiosensitization under the assumption that DSBs are the relevant lesions and that different pathways are involved in their repair. (A) outlines a first scenario in which DSBs are repaired by a heat sensitive and a heat resistant pathway. Survival curve A corresponds to the condition where all induced DSBs remain unrepaired and transformed to lethal lesions. This condition is approximated in yeast by several HRR mutants, but remains theoretical for cells of higher eukaryotes. Repair by the heat resistant repair pathway RP1 increases survival to the levels indicated by curve B, whereas repair by the heat sensitive repair pathway RP2 generates survival curve C. Exposure of wild type (WT) cells to heat will compromise RP2 and at the theoretical maximum of the effect will lead to cell survival according to curve B. This is equivalent to extensive radiosensitization. An RP2 mutant will show in the absence of heat the intrinsic radiosensitivity defined by survival curve B. Following exposure to heat the RP2 mutant will sustain no radiosensitization because RP1 is heat resistant. This scenario is implicit in the interpretation of results obtained with cells deficient in either D-NHEJ or HRR. (B) summarizes an alternative scenario in which DSBs are repaired by two heat-sensitive repair pathways; other details as in the first scenario. Exposure of wild type cells to heat will inhibit at the theoretical maximum of effect RP1 and RP2 and will result in survival curve A. This is equivalent to extensive radiosensitization. An RP2 mutant will have in the absence of heat the radiosensitivity defined by survival curve B and exposure to heat will cause significant radiosensitization and will generate at the theoretical maximum of the effect survival curve A. Thus, heat radiosensitization is observed as a result of the heat sensitivity of RP1 and will have a magnitude proportional to the contribution of RP1 to survival. It will be inaccurate to conclude here that RP2 is not involved in heat radiosensitization because the RP2 mutant can be radiosensitized by heat.
Similar conclusions could also be drawn assuming a scenario in which only one pathway (RP2) is involved in the repair of DSBs, or when mutants are examined under conditions compromising still active repair pathways. For reasons that are discussed below, the latter condition appears to have been unknowingly reached in the xrs-5 mutant when tested in the plateau phase.
An alternative scenario in which DSBs are repaired by two heat sensitive repair pathways is outlined in . In this case an RP2 mutant will show marked heat radiosensitization as a result of the response to heat of RP1. It would be inaccurate on the basis of this result to conclude that RP2 is not involved in heat radiosensitization.
The actual situation becomes more complex when RP1 and RP2 are differentially inhibited by heat, and when the contribution to DSB repair of the one repair pathway changes when the other is genetically inactivated. We present evidence below that both effects occur in mammalian cells.
Extending the picture to include three repair pathways, as it appears to be the case for DSB repair in higher eukaryotes (see below), will generate operational space for additional outcomes. The number of these potential outcomes will increase further if one considers different heat sensitivities among repair pathways and a dynamic equilibrium in their contribution to DSB repair that is disturbed when one of them is genetically inactivated. A complicating factor is also that often mutations only partially compromise a given repair pathway. This is particularly true of HRR due to its essential role in the survival of higher eukaryotes. On the basis of these arguments, as well as of those outlined below, we propose that the ‘simple’ interpretation given to heat radiosensitization data obtained with DNA DSB repair mutants has unjustifiably excluded DSBs from this phenomenon.
To be more specific, even if one considers only HRR and D-NHEJ in DSB repair, genetic inactivation of D-NHEJ will theoretically confine DSB repair to the activities of the remaining repair pathway, HRR. If this repair pathway is also sensitive to heat, then radiosensitization will be observed at levels commensurate to the inhibition by heat shock of this specific repair pathway and this effect can in no way be used as an argument that D-NHEJ is not a target of heat radiosensitization. This is particularly true when heat radiosensitization is quantified as the relative change in the survival of heated and irradiated cells as compared to cells exposed to radiation alone. Similar arguments can be developed for mutants defective in HRR. It will be even more difficult to justify such a ‘simple’ interpretation of mutant heat radiosensitization results when additional pathways of DSBs need to be considered (see below).
That heat shock has the capacity of inhibiting all active DSB repair pathways can be inferred from the pronounced effects of heat on all cellular metabolic activities. It can also be directly demonstrated by following DSB repair in cells exposed to heat. This is illustrated by the results shown in for plateau-phase CHO cells exposed to IR following heat exposures as indicated. In this experiment, a 30 min heat shock completely impairs DSB rejoining. This is an important observation as repair of DSBs is impressively robust and at present no genetic modifications are known capable of generating an effect even close to that shown here after exposure to heat (compare, for example, Citation[17]). Recall, for example, that cells defective in HRR show no detectable effect on DSB rejoining and that cells defective in D-NHEJ can repair the majority of IR-induced DSBs (see below). Even repair inhibitors cannot duplicate these effects, and treatment for example with wortmannin generates only a delay in DSB repair Citation[27]. Thus, among known physical or chemical agents or even among diverse genetic conditions, heat has the greatest potential of inhibiting DSB repair. It would certainly be surprising if this potential were not translated to increased radiosensitization.
Figure 5. Heat shock inhibits all pathways of DSB repair. (A) Repair kinetics of IR-induced DSBs in plateau-phase CHO cells exposed to 15 Gy X-rays after heating at 45.5°C for the indicated times. Repair of DSBs was measured by neutral filter elution. Note the complete inhibition of DSB rejoining that requires heating for about 30 min. Results are from Citation[19]. (B) Similar results obtained with the Ku80 deficient mutant xrs-5 Citation[19]. Note that in these cells, residual DSB repair, presumably carried out by repair pathways other than D-NHEJ, is already completely inhibited after 8 min of heating suggesting higher heat sensitivity. (C) Repair of DSBs in wild type, exponentially growing MEFs continuously incubated immediately after irradiation (20 Gy) at the indicated temperatures. Note the extensive inhibition of DSB rejoining observed under these conditions as well. Rejoining of DSBs was assayed here by pulsed-field gel electrophoresis. (D) Results similar to those of panel C obtained with LIG4−/− MEFs. Note that here again heat shock has a stronger effect on residual rejoining of DSBs as compared to wt cells.
![Figure 5. Heat shock inhibits all pathways of DSB repair. (A) Repair kinetics of IR-induced DSBs in plateau-phase CHO cells exposed to 15 Gy X-rays after heating at 45.5°C for the indicated times. Repair of DSBs was measured by neutral filter elution. Note the complete inhibition of DSB rejoining that requires heating for about 30 min. Results are from Citation[19]. (B) Similar results obtained with the Ku80 deficient mutant xrs-5 Citation[19]. Note that in these cells, residual DSB repair, presumably carried out by repair pathways other than D-NHEJ, is already completely inhibited after 8 min of heating suggesting higher heat sensitivity. (C) Repair of DSBs in wild type, exponentially growing MEFs continuously incubated immediately after irradiation (20 Gy) at the indicated temperatures. Note the extensive inhibition of DSB rejoining observed under these conditions as well. Rejoining of DSBs was assayed here by pulsed-field gel electrophoresis. (D) Results similar to those of panel C obtained with LIG4−/− MEFs. Note that here again heat shock has a stronger effect on residual rejoining of DSBs as compared to wt cells.](/cms/asset/ace5ad20-b803-4e7c-a0ae-33961d1dee93/ihyt_a_278504_f0005_b.gif)
While nearly 30 min of heat exposure are required for the complete inhibition of DSB rejoining in repair proficient cells such as CHO (), 8 min suffice for an equivalent effect in the Ku80 deficient xrs-5 mutant. This is direct evidence that in this mutant residual DSB repair carried by pathways other than D-NHEJ is more heat sensitive than D-NHEJ itself, and provides experimental support for differential heat sensitivity of the various DSB repair pathways (see above). Results similar to those obtained with Chinese hamster cells have also been recently obtained using wild type MEFs and their LIG4−/− counterparts (). Here again extensive inhibition of DSB rejoining is observed that is more pronounced in the D-NHEJ mutant. While the nature of the repair pathways operating in cells deficient in D-NHEJ is reviewed next, it is important to conclude from these results that heat has the potential of inhibiting all DSB repair irrespective of the pathway by which it is mediated.
Backup pathways of NHEJ in heat radiosensitization
While all available mutants of D-NHEJ show clear impairment in their capacity to repair DSBs, the defect does not manifest as a complete absence of DSB repair. Notably, these mutants repair the majority of IR-induced DSBs albeit with slow kinetics. This has been documented in several reports Citation[17], Citation[27], Citation[28], Citation[29], Citation[30], Citation[31] and is also illustrated in with the results of LIG4−/− MEFs. Nearly 75% of IR-induced DSBs could be repaired within 8 h under the conditions used, and it is likely that longer repair times would have allowed further repair to take place. These observations demonstrate a plasticity among DSB repair pathways in the sense that inactivation of one repair pathway allows another repair pathway, operating as a backup, to take over. As a result, a backup repair pathway handles in a mutant a larger fraction of DSBs than it normally handles in the wild type cells. This observation suggests that pathway selection may not be made based on the character or type of the IR-induced DSB, as these characteristics are not expected to change in the mutant as compared to the wild type cells. Also, the fact that these backup pathways are particularly heat sensitive (see ) generates strong expectations for persistent radiosensitization. But what is the nature of these pathways of DSB repair that remain active when D-NHEJ is inhibited
The first interpretation for the D-NHEJ independent DSB repair activity was that it reflects HRR. It was therefore surprising to find that cells with mutations in genes implicated in HRR do not show the expected repair defect and that even a double DT40 Ku70−/−/Rad54−/− mutant repairs DSBs with an efficiency practically identical to that of the Ku70−/− mutant Citation[16], Citation[17], Citation[29]. The hypothesis was therefore advanced for the operation of alternative DSB repair pathways, distinct from D-NHEJ and HRR that operate as backup. This hypothesis found additional support in a series of biochemical and genetic investigations which suggested that this form of DSB repair is a distinct form of end joining, which is normally suppressed by D-NHEJ Citation[32]. We proposed the term B-NHEJ for this form of NHEJ to facilitate differentiation from D-NHEJ and to emphasize its putative backup function Citation[16].
Recent work implicates DNA Ligase III in B-NHEJ Citation[33], Citation[34] and suggests that the repair module PARP-1/XRCC1/DNA Ligase III Citation[28], Citation[34], which is firmly implicated in the repair of single strand breaks (SSB) Citation[35], also contributes to DSB repair. This repair module is included in as an alternative pathway of DSB repair, in addition to the D-NHEJ and HRR. By increasing the number of pathways available for DSB repair from two to three we enter the expanded space of combinations for the interpretation of heat radiosensitization results outlined under the second scenario in .
The operation of alternative pathways of NHEJ has also been suggested by experiments evaluating joining of ends generated in naked DNA in vitro and transfected into cells for processing. Mammalian cells demonstrate an extraordinary ability to join such transfected DNA, either by direct ligation or by utilizing microhomologies Citation[36], Citation[37]. Notably, cells deficient in DNA-PKcs Citation[38], Citation[39], Citation[40], Ku Citation[40], Citation[41], XRCC4 Citation[40], Citation[41] or DNA ligase IV Citation[40] show high potential of end joining with preferential use of microhomologies Citation[40], Citation[41]. This microhomology dependent end joining may overlap partly or completely with B-NHEJ and has been recently shown to be involved in the repair of DNA breaks created during assembly of antigen receptor genes Citation[42], Citation[43], Citation[44], Citation[45]. These developments provide solid evidence for the acute biological significance of the backup pathway of DSB repair and implicate it in the chromosomal translocations of lymphoid cancers.
The above outline and the properties of B-NHEJ offer a useful framework for the analysis of the xrs-5 heat radiosensitization results () which appeared to divert from results obtained with similar mutants (see above). We recently reported that while D-NHEJ is only marginally modulated as exponentially growing cells progress into the plateau phase of growth, B-NHEJ is markedly compromised Citation[30]. This reduction in B-NHEJ activity is of marginal consequence in repair proficient cells where D-NHEJ is active, but becomes important in D-NHEJ deficient cells that rely extensively on B-NHEJ for DSB repair. This is illustrated in , where no significant modification in DSB repair capacity is noted when repair proficient cells progress from the logarithmic to the plateau phase of growth. On the other hand, in LIG4−/− MEFs that are compromised in D-NHEJ and rely extensively on B-NHEJ, a marked reduction in end joining efficacy is observed as cells enter the plateau phase of growth Citation[30]. We have since reproduced this observation in a variety of D-NHEJ deficient mutants and demonstrated its general validity.
Figure 6. Rejoining of DSBs in exponentially growing (2d) and plateau phase (4d) MEFs obtained either from wild type or LIG4−/− animals. Cells were exposed to 20 Gy and analysed for DSB repair by pulsed-field gel electrophoresis Citation[30]. Note the reduction in repair capacity of LIG4−/− MEFs as they enter the plateau phase of growth.
![Figure 6. Rejoining of DSBs in exponentially growing (2d) and plateau phase (4d) MEFs obtained either from wild type or LIG4−/− animals. Cells were exposed to 20 Gy and analysed for DSB repair by pulsed-field gel electrophoresis Citation[30]. Note the reduction in repair capacity of LIG4−/− MEFs as they enter the plateau phase of growth.](/cms/asset/fa92b4bf-2f44-42ba-aa93-8eca606c709d/ihyt_a_278504_f0006_b.gif)
The impairment of B-NHEJ as cells stop growing and enter a plateau phase is relevant for understanding the xrs-5 data shown in . Since plateau phase cells were used in the experiments, B-NHEJ was probably compromised. In addition xrs-5 cells are deficient in D-NHEJ and under the conditions used also compromised for HRR – because in the plateau phase, cells were predominantly in the G1 phase of the cell cycle where HRR may not be active (see above). Thus, all three presently known pathways of DSB repair were moderately to severely compromised, which brought the system close to the condition of no residual repair as discussed for the first scenario in . As a result, heat radiosensitization was substantially reduced. When experiments are carried out with exponentially growing D-NHEJ mutants, or with mutants showing partial defects, ample residual heat radiosensitization is observed as a result of inhibition of B-NHEJ and HRR, both of which will be active under these conditions.
Implications of an upper limit of heat radiosensitization
The question as to whether an upper limit exists in heat radiosensitization has been raised early on in the field and has been reviewed Citation[46]. Experiments reported by different investigators support the view that an upper limit is approached in radiosensitization after heating at a given temperature for increasing periods of time. After this limit is reached, longer heating enhances heat-mediated lethality, but does not produce further radiosensitization. A limit in heat radiosensitization suggests the existence of a defined class of lesions whose repair can be affected by heat. It will be essential for the molecular understanding of heat radiosensitization to characterize this class of lesions and the interplay between the pathways involved in their repair.
A limit in heat radiosensitization is indicated by the results shown in for CHO cells, which together with the results of xrs-5 cells allow the formulation of models regarding type of lesions involved and repair pathways employed. Simple visual inspection of the survival curves obtained with the two types of cells indicates large differences in radiosensitivity in cells irradiated without pre-heating, but only marginal differences in radiosensitivity following a 30–40 min pre-exposure to heat. This trend is better illustrated in , which summarizes the same data plotted as D1 (radiation dose required for 1% survival) as a function of the survival measured following exposure to heat alone (a function of heat duration and intrinsic heat sensitivity) Citation[18]. In CHO cells, D1 rapidly declines with increasing heating effect, but the change becomes progressively smaller and approximates asymptotically the D1 values of the xrs-5 mutant. Notably, there is only a small reduction in D1 in xrs-5 cells.
Figure 7. Heat radiosensitization has an upper limit. (A) D1, the radiation dose required to reduce survival to 1%, in plateau phase CHO and xrs-5 cells as a function of the resulting cell survival in cells exposed to heat alone. Results obtained from Citation[18]. (B) Survival of exponentially growing CHO cells exposed either to 4 MeV X-rays, or to accelerated 12C ions. Irradiation was carried out after incubation for 1h either at 37°C or at 43°C. Note the absence of heat radiosensitization after exposure to the high LET carbon ions Citation[47].
![Figure 7. Heat radiosensitization has an upper limit. (A) D1, the radiation dose required to reduce survival to 1%, in plateau phase CHO and xrs-5 cells as a function of the resulting cell survival in cells exposed to heat alone. Results obtained from Citation[18]. (B) Survival of exponentially growing CHO cells exposed either to 4 MeV X-rays, or to accelerated 12C ions. Irradiation was carried out after incubation for 1h either at 37°C or at 43°C. Note the absence of heat radiosensitization after exposure to the high LET carbon ions Citation[47].](/cms/asset/7130d557-1b4f-4003-8904-dc875279ba52/ihyt_a_278504_f0007_b.gif)
The observation that the radiosensitivity of repair proficient cells approaches after sufficient heat exposure that of a DSB repair-deficient mutant is interesting for several reasons. First, it suggests that heat radiosensitization in repair proficient CHO cells derives from inhibition of all repair pathways that are deficient in the xrs-5 mutant under the conditions tested, i.e. D-NHEJ, B-NHEJ and HRR (see above). Second, because the number of lethal events scored (see for details) at the maximum of heat radiosensitization is smaller than the number of DSBs induced by IR (20–40 per Gy), it follows that only a subset of DSBs contributes to the effect. Additional work will be required to establish whether stochastic processes, or other biological and biochemical characteristics, define this subset of DSBs. In any event the results shown allow the conclusion that this subset of DSBs will utilize all repair pathways that are compromised in the xrs-5 mutant.
Additional evidence for a limit in heat radiosensitization comes from the cell cycle studies summarized in . In these experiments cells irradiated at mitosis were extremely radiosensitive and showed no evidence for heat radiosensitization Citation[1]. Thus, even in the absence of any DSB repair compromising mutation, conditions are generated in the cell cycle impairing DSB repair (probably by enhancing misrepair), and this condition compromises heat radiosensitization. This implies that the radiosensitization observed in other phases of the cell cycle derives from inhibition in the repair of those DSBs, the repair of which is naturally impaired at mitosis. We have previously reviewed available genetic evidence regarding the source of the cell cycle dependent fluctuations in cell radiosensitivity to killing and presented arguments for HRR as an important contributor to this effect Citation[10]. Extending these arguments to heat radiosensitization, and considering the lack of radiosensitization of mitotic cells, it can be postulated that inhibition of HRR is an important component of heat radiosensitization.
A further set of experiments indicating a limit in heat radiosensitization and allowing inferences on the nature of the DSBs involved in the effect has been carried out with high LET radiation. Results of these experiments are shown in and indicate that while significant heat radiosensitization is observed after exposure of cells to low LET radiation, only marginal radiosensitization is seen after exposure to 12C ions near the Bragg peak Citation[47]. Notably, the radiation sensitivity of heated cells is very similar regardless of the type of radiation used and close to that of untreated cells exposed to high LET radiation (after correcting for the effect of heat alone).
DSBs versus base damage in heat radiosensitization
While the arguments developed above focus on DSBs, they by no means exclude in principle other types of lesions such as base damage as potential contributors to heat radiosensitization Citation[25], Citation[26]. They were developed for the purpose of generating a picture of heat radiosensitization that accommodates existing results and analyses them in the light of recent developments in the field. Central purpose is to develop alternative interpretations for heat radiosensitization results obtained with DSB repair deficient mutants and to indicate that the arguments used to rule out DSBs as targets of heat radiosensitization need to be reconsidered. Our working hypothesis remains therefore that DSBs and the inhibition of their processing is the key determinant of heat radiosensitization. What then about base damage?
Base damage can certainly contribute to heat radiosensitization and arguments to this end have been developed and discussed Citation[25], Citation[26]. However, in our view, the extent of such contribution to heat radiosensitization will be proportional to the contribution of this lesion to cell killing in non-heated cells. Since this contribution remains undefined and is likely to be small, a conclusive evaluation of a contribution to heat radiosensitization is not possible at this time. If inhibition in the processing of base damage were solely responsible for heat radiosensitization, it is not clear why mitotic cells should show no heat radiosensitization.
Also, the argument that base damage contributes to heat radiosensitization via its indirect transformation to DSBs Citation[25], Citation[26] cannot withstand scrutiny since DSBs are processed by the same repair pathways () regardless of origin. Only the selection of the repair pathway will be biased by the context of their production. Thus, the same arguments and conclusions developed for directly induced DSBs should be applied to those induced indirectly. If one assumes that DSBs are not involved in heat radiosensitization when they are induced directly, they cannot be invoked to explain heat radiosensitization when they are induced indirectly.
Conclusions
On the basis of the above outline we derive a set of tentative conclusions as working hypotheses for further work:
Heat radiosensitizes predominantly by inhibiting the processing of DSBs.
All three pathways involved in the repair of DSBs contribute to the overall radiosensitization. Their relative contribution to radiosensitization will depend upon their relative contribution to DSB repair under the experimental conditions employed and upon their individual heat sensitivity. While the mechanisms of inhibition by heat of HRR and B-NHEJ remain to be characterized, inhibition of D-NHEJ may at least in part be caused by inactivation of Ku Citation[48], Citation[49].
While heat has the potential of inhibiting repair of all DSBs induced by IR, it cannot transform all IR DSBs to lethal lesions. Only a subset of DSBs induced by low LET radiation has the characteristics required for heat radiosensitization. Susceptibility to misrepair may be a key characteristic of this subset.
The contribution of base damage to heat radiosensitization is expected to be proportional to the contribution of this lesion to killing after exposure to IR alone, and in general small.
Acknowledgements
Work was supported by P01-CA75556 awarded by NCI, NIH, DHHS, as well as by grants from the German Research Society (DFG) and Federal Ministry for Education and Research (BMBF).
References
- Dewey WC, Sapareto SA, Betten DA. Hyperthermic radiosensitization of synchronous Chinese hamster cells: Relationship between lethality, & chromosomal aberrations. Radiat Res 1978; 76: 48–59
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 2002; 66: 630–670
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002; 23: 687–696
- Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 2003; 22: 5792–5812
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4: 1–11
- Wyman C, Ristic D, Kanaar R. Homologous recombination-mediated double-strand break repair. DNA Repair 2004; 3: 827–833
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews 1999; 63: 349–404
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 2000; 19: 3398–3407
- Jasin M. Homologous repair of DNA damage and tumorigenesis: The BRCA connection. Oncogene 2002; 21: 8981–8993
- Tamulevicius P, Wang M, Iliakis G. Homology-directed repair is required for the development of radioresistance during S phase: Interplay between double-strand break repair and checkpoint response. Radiat Res 2007; 167: 1–11
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res 2002; 509: 49–78
- Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003; 4: 712–720
- Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 2003; 194: 77–95
- Rothkamm K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23: 5706–5715
- Metzger L, Iliakis G. Kinetics of DNA double strand breaks throughout the cell cycle as assayed by pulsed field gel electrophoresis in CHO cells. Int J Radiat Biol 1991; 59: 1325–1339
- Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res 2004; 104: 14–20
- Wang H, Zeng Z-C, Bui T-A, Sonoda E, Takata M, Takeda S, et al. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene 2001; 20: 2212–2224
- Iliakis G, Seaner R. A DNA double strand break repair deficient mutant of CHO cells shows reduced radiosensitization after exposure to hyperthermic temperatures. Int J Hyperthermia 1990; 6: 801–812
- Iliakis G, Seaner R, Okayasu R. Effects of hyperthermia on the repair of radiation induced DNA single and double strand breaks in DNA double strand break repair deficient and repair proficient cell lines. Int J Hyperthermia 1990; 6: 813–833
- Raaphorst GP, Maude-Leblanc J, Li L. Evaluation of recombination repair pathways in thermal radiosensitization. Radiat Res 2004; 161: 215–218
- Dynlacht JR, Bittner ME, Bethel JA, Beck BD. The non-homologous end-joining pathway is not involved in the radiosensitization of mammalian cells by heat shock. J Cell Physiol 2003; 196: 557–564
- Raaphorst GP, Thakar M, Ng CE. Thermal radiosensitization in two pairs of CHO wild-type and radiation-sensitive mutant cell lines. Int J Hyperthermia 1993; 9: 383–391
- Yin HL, Suzuki Y, Matsumoto Y, Tomita M, Furusawa Y, Enomoto A, et al. Radiosensitization by hyperthermia in the chicken B-lymphocyte cell line DT40 and its Derivatives lacking nonhomologous end joining and/or homologous recombination pathways of DNA Double-strand break repair. Radiat Res 2004; 162: 433–441
- Kampinga HH, Kanon B, Konings AWT, Stackhouse MA, Bedford JS. Thermal radiosensitization in heat- and radiation-sensitive mutants of CHO cells. Int J Radiat Biol 1993; 64: 225–230
- Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (>43°C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 2004; 20: 131–139
- Kampinga HH, Dikomey E. Hyperthermic radiosensitization: Mode of actions and clinical relevance. Int J Radiat Biol 2001; 77: 399–408
- Wang H, Zhao-Chong Z, Perrault AR, Cheng X, Qin W, Iliakis G. Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res 2001; 29: 1653–1660
- Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 2006; 34: 6170–6182
- DiBiase SJ, Zeng Z-C, Chen R, Hyslop T, Curran WJ, Jr., Iliakis G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res 2000; 60: 1245–1253
- Windhofer F, Wu W, Wang M, Singh SK, Saha J, Rosidi B, et al. Marked dependence on growth state of backup pathways of NHEJ. Int J Radiation Oncology Biol Phys 2007; 68: 1462–1470
- Windhofer F, Wu W, Iliakis G. Low Levels of DNA Ligases III and IV Sufficient for Effective NHEJ. J Cell Physiol 2007; 213(2)475–483
- Perrault R, Wang H, Wang M, Rosidi B, Iliakis G. Backup pathways of NHEJ are suppressed by DNA-PK. J Cell Biochem 2004; 92: 781–794
- Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res 2005; 65: 4020–4030
- Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 2004; 279: 55117–55126
- Caldecott KW. Mammalian DNA single-strand break repair: An X-ra(y)ted affair. Bioessays 2001; 23: 447–455
- Roth DB, Porter TM, Wilson JH. Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol 1985; 5: 2599–2607
- Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: Role for short sequence homologies in the joining reaction. Mol Cell Biol 1986; 6: 4295–4304
- Chang C, Biedermann KA, Mezzina M, Brown JM. Characterization of the DNA double strand break repair defect in scid mice. Cancer Res 1993; 53: 1244–1248
- Harrington J, Hsieh CL, Gerton J, Bosma G, Lieber MR. Analysis of the defect in DNA end joining in the murine scid mutation. Mol Cell Biol 1992; 12: 4758–4768
- Verkaik NS, Esveldt-van Lange REE, van Heemst D, Brüggenwirth HT, Hoeijmakers JHJ, Zdzienicka MZ, et al. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur J Immunol 2002; 32: 701–709
- Kabotyanski EB, Gomelsky L, Han J-O, Stamato TD, Roth DB. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res 1998; 26: 5333–5342
- Soulas-Sprauel P, Le Guyader G, Rivera-Munoz P, Abramowski V, Olivier-Martin C, Goujet-Zalc C, et al. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J Exp Med 2007; 204: 1717–1727
- Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong S-Y, et al. Rag mutations reveal robust alternative end joining. Nature 2007; 449: 483–486
- Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell 2007; 131: 223–225
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007; 449: 478–482
- Hahn GM. Hyperthermia and cancer. Plenum Press, New York 1982
- Gerner EW, Leith JT. Interaction of hyperthermia with radiations of different linear energy transfer. Int J Radiat Biol 1977; 31: 283–288
- Burgman P, Ouyang H, Peterson S, Chen DJ, Li GC. Heat inactivation of Ku autoantigen: Possible role in hyperthermic radiosensitization. Cancer Res 1997; 57: 2847–2850
- Matsumoto Y, Suzuki N, Sakai K, Morimatsu A, Hirano K, Murofushi HA. Possible mechanism for hyperthermic radiosensitization mediated through hyperthermic lability of Ku subunits in DNA-dependent protein kinase. Biochem Biophys Res Commun 1997; 234: 568–572