Other contributors to the report: T. Akimoto (Gunma, Japan), M. Arimura (Nagasaki, Japan), K. Ariyoski (Nagasaki, Japan), J. Bull (Houston, USA), Y. Harima (Osaka, Japan), T. Hasegawa (Osaka, Japan), M. Hiraoka (Kyoto, Japan), N. Huilgol (Mumbai, India), M. Hurwitz (Boston, USA), I. Ichikawa, (Kyoto, Japan), H. Imada (Kitakyushu, Japan), Y. Itoh (Aichi, Japan), K. Katayama (Fukui, Japan), K. Kishida (Kyoto, Japan), N. Kobayashi (Nagasaki, Japan), S. Kodama (Osaka, Japan), S. Kokura (Kyoto, Japan), T. Kondo (Toyama, Japan), M. Kosaka (Toyota, Japan), C. Lee (Minneapolis, USA), S. Malta (Verona, Italy), N. Matsuda (Nagasaki, Japan), T. Matsuda (Tokyo, Japan), H. Matsumoto (Fukui, Japan), Y. Matsumoto (Tokyo, Japan), M. Miyazawa (Osaka, Japan), M. Morimoto (Nagasaki, Japan), J. Nagao (Kumamoto, Japan), I. Nishide (Osaka, Japan), K. Ohnishi, (Nara, Japan), K. Ohtsuka, (Aichi, Japan), T. Otsuka (Aichi, Japan), K. Okamura (Okayama City, Japan), K. Ono (Osaka, Japan), S. Osinsky (Kiev, Ukraine), H. Sahinbas (Bohum, Germany), H. Sakurai (Gunma, Japan), S. K. Shrivastava (Mumbai, India), K. Suzuki (Nagasaki, Japan), M. Suzuki (Nagasaki, Japan), A. Takahashi (Nara, Japan), Y. Tanaka (Tokyo, Japan), Y. Tanaka (Osaka, Japan), H. Terashima (Fukuoka, Japan), H. Terunuma (Tokyo, Japan),. A. Toki (Tama, Japan), R. U (Raleigh, USA), H. Ueda (Wakayama, Japan), K. Ueda (Aichi, Japan), K. Watanabe (Nagasaki, Japan), G. Yamamoto (Osaka, Japan), M. Yamauchi (Nagasaki, Japan), S. Yukawa (Osaka, Japan)
Preface
TSUTOMU SUGAHARA1 & CHANG W. SONG2
1Japan Health Foundation, Kyoto and 2University of Minnesota, Minneapolis, USA
It has been more than three decades since laboratory experiments demonstrated that heating at 42°–45°C kills cancer cells, including radioresistant hypoxic cancer cells, sensitises cancer cells to ionising radiation or certain chemotherapy drugs, and preferentially destroys blood vessels in tumours relative to that in normal tissues. Furthermore, human normal cells at confluent stage in vitro were demonstrated to be considerably heat-resistant when compared to neoplastic cells. Based on these exciting observations, ‘hyperthermia’ or ‘thermotherapy’ emerged as a new and promising cancer treatment regimen and was quickly embraced by the oncology community, particularly by radiation oncologists. However, despite the compelling biological rationale, the enthusiasm for clinical application of hyperthermia considerably waned in recent years mainly because it was realised that heating human tumours, especially bulky and deep-seated tumours, to cytotoxic temperatures, i.e. 42°–45°C, is rather difficult or practically impossible. Nonetheless, in a number of recent clinical trials, heating human tumours at mild temperatures, i.e. 39°–42°C which is suboptimal for causing direct cell killing or tumour vascular damage, was still found to be effective in enhancing the tumour response to radiotherapy or chemotherapy. Recent studies with rodent tumours or clinical studies indicated that such an enhancement of radiotherapy or chemotherapy of tumours by mild temperature heating is most likely due to an increase in blood perfusion and a resultant improvement in tumour oxygenation. These developments imply that 42°–45°C is not the only temperature range useful for the improvement of cancer control by hyperthermia.
Indeed, it is increasingly evident that mild temperature hyperthermia (MTH) at 39°–42°C has an important role in the hyperthermia arsenal beside other anti-cancer heating strategies including moderate temperature hyperthermia at 42°–45°C, ablation therapy using temperatures higher than 50°C as well as fever-range whole body hyperthermia (FR-WBH). An analogy of the optimum use of multiple weaponry derives from old Japanese history. During the ‘Shogun’ era in Japan, some samurais (warriors) favoured ‘Nitoryu’, meaning ‘a way of fighting using a pair of short and long swords’. The strongest samurai in Japanese history was probably Mr Musashi Miyamoto (1584–1645), who mastered ‘Nitoryu’ and used both short and long swords depending on the enemy and situation. Likewise, in treating various human diseases with thermotherapy, a time-temperature profile should be rationally chosen depending on the situation in order to maximally exploit various cellular, physiological and immunological effects of heat shock.
In order to discuss recently revealed, clinically relevant biological effects of heat shock as well as to review recent clinical results of hyperthermic treatment of human tumours, ‘The Kadota Fund International Forum 2004’ was held in June 14–18, 2004, at Awaji Yumebutai International Conference Centre, Awaji Island, Hyogo, Japan with generous support from the Japan Health Foundation, Kadota Fund and Hyogo International Association. A total of 68 clinicians and biologists from Japan and elsewhere were invited to the Forum. In this two-part report, ‘Clinical Aspects of Hyperthermia’ and ‘Biological Aspects of Hyperthermia’, the subjects discussed, conclusions and recommendations made at the Forum are reported.
It must be stressed that there were some disagreements amongst the participants on certain subjects discussed at the Forum. Consequently, the contents of this report do not necessarily represent the opinions of all participants. Likewise, many investigators in the thermotherapy community who did not participate in the Forum may dissent from the conclusions reported herein. Nevertheless, we hope this report may stimulate exchange of different views among the investigators for the further improvement of application of thermotherapy not only for the treatment of cancers but also other diseases. Finally, it should be noted that important subjects in thermotherapy such as hyperthermia physics/engineering and thermal ablation therapy were not included in the Forum due to space and budgetary limitations.
Part II. A. Cell biological aspects of hyperthermia
HARM H. KAMPINGA
Faculty of Medical Sciences, Department of Radiation and Stress Cell Biology, Division of Cell Biology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Introduction
At the time of the consensus report of 1990 Citation[1], optimistic ideas were put forward regarding the potential selective heat sensitivity of tumour versus non-tumour cells, the development of predictive assays in hyperthermia, and the heat-induced improvement of responses to radiotherapy and/or chemotherapy on the basis of mechanistic information from in vitro experiments. Although none of these ideas turned out to be realistic, still a lot of progress was made regarding the cell biological aspects of hyperthermia.
Although several key-players in the recent development of new insights into hyperthermic cell biology were not present at the Forum, attempts were made by the biologist present at the meeting to include the work of others in this report, trying to be as objective and complete as possible. The cell biology report describes the effects of heat alone or combined with radiation or chemotherapeutic agents. Special attention was given to the regulation and function of heat shock proteins (HSPs) and to the various forms of cell death expression after a clinically relevant heat shock.
Protein damage as molecular cause for the biological effects of heat
As was recognised in the early 1970s, protein damage is the main molecular event underlying the biological effects of hyperthermia in the clinically relevant temperature range of 39°–45°C Citation[1]. At these temperatures, direct damage to DNA does not seem to be involved in cell killing by heat; the activation energies for direct DNA damage are in the range of 25–35 kcal/mol and thus unrelated to the ΔH of heat killing (±140 kcal/mol). Yet, DNA damage may be induced indirectly via protein damage and indeed depurinations in DNA have been observed to occur with comparable activation energies as those for heat killing Citation[2]. As recent studies Citation[3] suggest that there is a relation between immunohistological markers of DNA-double strand breaks (DSB) and heat toxicity, the role of (indirect) DNA damage will be discussed separately below.
Besides proteins, lipids are also known to be affected (fluidity) by heat. Yet, research of the last 15 years has revealed that lipid damage is reversible and not directly causative for heat killing, although lipid modification may modulate the magnitude of biological effects of heat indirectly by influencing (accelerating/decelerating) the rate of protein aggregation Citation[4].
The notion that protein damage plays a central role in the biological effects of heat is based on a variety of different experimental evidence:
The activation energy for protein denaturation is in the range of 80–500 kcal/mol which is within the range for heat killing Citation[5].
Biophysical approaches, especially those from the group of (the late) James Lepock such as differential scanning calorimetry Citation[6], electron spin resonance Citation[7], as well as work with model proteins Citation[8–10] have directly shown that substantial protein denaturation occurs in the clinically relevant temperature range (reviewed in Citation[11]).
Biochemical and cell fractionation experiments have shown that (as a result of protein denaturation) proteins become insoluble and aggregate, thereby affecting many macromolecular structures and their function(s). Protein aggregation, as indicated by the increase in detergent-insoluble material, is probably the best parameter correlating protein damage to the extent of cell death Citation[10], Citation[12].
Many modulators of heat-induced cell death (chemical sensitisers, protectors, or heat shock proteins) act via affecting the rate of protein denaturation, aggregation or disaggregation Citation[10–16].
Beside direct thermal denaturation of proteins, heat treatments (especially mild ones) also increase the metabolic rate of cells. This, in turn, increases the production of free radicals and indirectly will lead to damage, especially to proteins. This could explain the link between oxidative stress, heat shock and heat shock proteins.
As mentioned above, it was recently suggested that (indirect) DNA damage may also contribute to heat-induced lethality Citation[3]. This was based on the observation that heat shock induces so-called γ-H2AX foci. H2AX, a member of the histone H2A family, is rapidly phosphorylated in response to ionising radiation and forms nuclear foci that are quantitatively related to the number of radiation-induced DNA DSB Citation[20]. It was hypothesised Citation[3] that DNA depurination (indirect, as a result of protein damage) and delayed repair of base damage after heat shock Citation[6] results in the formation of DNA DSB, which may contribute to cell lethality after heat shock. Although no complete consensus was reached regarding the role of DNA DSB in heat-induced killing, a number of arguments against this hypothesis were raised:
Many biochemical/biophysical approaches have failed to detect DSB after heat alone. It can be argued that this is a matter of sensitivity: γ-H2AX foci are readily detectable after irradiation with doses as low as 0.5 Gy whilst most other assays require 2 Gy or higher. However, heat treatments at thermal doses equivalent to >5 Gy in inducing γ-H2AX foci were never found to induce DSB when determined with other methods such as pulsed field gel electrophoresis that are able to detect low levels of damage.
BrdUrd incorporation, known to destabilise DNA and to enhance radiosensitivity, does not enhance heat killing Citation[21].
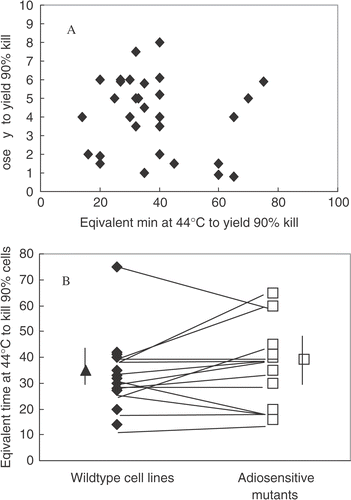
No correlation exists between heat sensitivity and radiosensitivity () and more specifically, in isogenic panels of cells pro- or deficient in DNA DSB repair no general trend is seen towards a thermal hypersensitivity of the DSB repair deficient, radiosensitive cells ().
Hyperthermia does not induce chromosomal aberrations except in S-phase cells Citation[22]. In S-phase cells, chromatid type damage is seen after (relatively severe) heat treatments but this can be explained by thermal inhibition of DNA synthesis (correlated to protein aggregation) and the resulting increased single-strandedness of the DNA Citation[22]. Heating also does not cause cell transformation, unlike radiation.
Figure 1. Absence of a correlation between radiation sensitivity and heat sensitivity. Panel A shows a cross-correlation of radiation sensitivity, expressed as the dose of X-ray required to kill 90% of the cells, with heat sensitivity, expressed as the equivalent time of heating at 44°C required to kill 90% of the cells, in 30 different mouse cell lines derived from the literature. Panel B shows the comparison of heat sensitivity between various radiosensitive mutants (deficient in either non-homologous end-joining or in homologous recombination) and their isogenic repair proficient counterparts. The average sensitivity of the groups (indicated by the single points with error bars) is not different, nor is there a trend for increase or decrease in heat sensitivity in matched panels (indicated by the lines).
Regulation of the heat shock response
One of the most revolutionary developments in the field of heat shock in the last 15 years is the unravelling of the heat shock response, i.e. the control mechanism involved in the induction of expression of the HSP-genes. It has been known since the early 1960s that temperature elevations transiently upregulate members of the heat shock gene family Citation[23] that were later found to encode the class of proteins we now know as heat shock proteins Citation[24]. The upregulation of these HSPs results in a transient resistance of cells towards heat shock (thermotolerance) and a variety of other stresses Citation[25] although thermotolerance can also arise without elevation of Hsp levels. In such a Hsp-synthesis independent thermotolerance HSPs may still play a role; they may be redistributed to essential (thermolabile) sites in the cells and as such provide protection.
The mechanism responsible for the heat shock response is an autoregulatory loop in which a specific heat shock transcription factor (HSF-1) plays a central role Citation[26]. Under non-stressful conditions, constitutively synthesised heat shock proteins (Hsp90, Hsp70) bind monomeric HSF-1 to keep it inactive and in the cytosol. This binding is dynamic and upon stress, the equilibrium between bound and released HSF-1 is shifted towards the released form, because the HSPs are now binding to denatured proteins for which they have a higher affinity. Therefore, protein damage can be considered to be a trigger that is responsible for HSF-1 activation. Upon release from the HSP, HSF-1 enters the nucleus, forms trimers, and binds to specific regulatory elements present on all heat shock genes, so-called heat shock elements (HSE). The HSF-1 molecules are then phosphorylated and activate transcription of the heat shock genes leading to the elevated expression of HSP. Attenuation of the inducible transcriptional response involves dissociation of the HSF-1 trimer and loss of activity. Dissociation involves a factor named heat shock factor binding protein 1 (HSPB1) that negatively affects HSF-1 DNA-binding activity in an Hsp70-dependent manner. How HSF-1 is converted from its trimeric to its monomeric form is not well understood, but it is thought that the increased levels of HSP assure that HSF-1 is maintained again in its inert monomeric form after this conversion. HSBP1 may play a highly intriguing role in this conversion as it was shown to inhibit the induction of HSP-expression Citation[27]. Alterations of the level of HSBP1 expression in Caenorhabditis elegans exerted severe effects on survival of the animals after thermal and chemical stress Citation[27]. Therefore, HSBP1 might be a therapeutic target to interfere with the cellular ability to induce thermotolerance. Also, screen for pharmacological agents that affect HSF-1 or its activation seems worthwhile as they may have future clinical application to enhance heat sensitivity and/or prevent the induction of thermotolerance.
Molecular chaperones
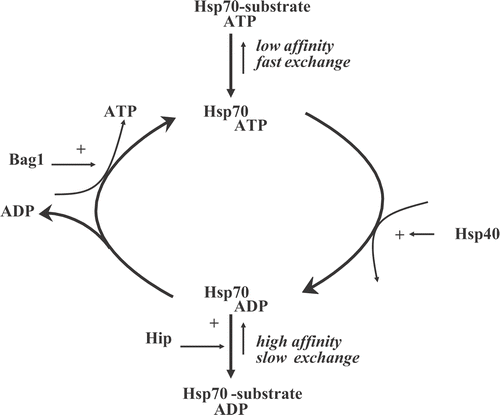
In addition to the steady increase in the understanding of the induction of the heat shock response, our understanding of the regulation and function of heat shock proteins has also greatly increased. Since the early 1980s, it was discovered that HSPs belong to the super family of proteins called molecular chaperones. Molecular chaperones are defined as proteins that bind to non-native or (partially) unfolded proteins and assist in their correct assembly by preventing their non-productive association (aggregation) with other proteins Citation[28]. Through binding and release cycles, unfolded proteins can slowly acquire their final conformation (). If released without being completely folded, a protein will re-enter the cycle until it reaches native structure. Under non-stressful circumstances, chaperones are involved in many physiological processes in which proteins are on their way to their native structure (translation) or during which they need to be in a partially unfolded state (during transport over membranes or during assembly into multiprotein complexes) Citation[29].
Figure 2. Reaction cycle of the Hsp70 chaperone machine and its regulation by cofactors.
In mammalian cells at least four main classes of HSP chaperones can be distinguished: a) small HSPs; b) chaperonins (Hsp60 in mitochondria, TCP-1 in the cytosol); c) the Hsp70 machine; d) the Hsp90 machine. All of these main chaperones have both constitutively expressed isoforms (controlled by other factors besides HSF-1 alone) as well as strongly heat-inducible family members (mainly HSF-1 dependent). Although not fully clear yet, each of these main chaperones appears to have specific substrates or act in specific steps of the folding pathway of a protein.
Of particular interest are the Hsp90 substrates which include a wide variety of signal-transducing proteins that regulate cell growth and differentiation, such as protein kinases and steroid hormone receptors Citation[30]. Since these Hsp90 client proteins play important roles in the regulation of the cell cycle, cell growth, cell survival, apoptosis, and oncogenesis, HSP90 inhibitors may be potential and effective cancer chemotherapeutic drugs. Indeed, earlier (phase I) trials have shown encouraging results with the Hsp90 inhibitors geldanamycin and especially with its derivative 17-allylaminogeldanamycin (17-AAG), that appears to have lower toxicity Citation[31].
In cell-free systems, all of the main chaperones can bind to unfolded proteins, prevent their irreversible aggregation, thus maintaining them in a refolding competent state (i.e., they can be reactivated to an active protein upon stress relief). However, the actual ‘reactivation’ step usually requires the action of the main chaperone system: the Hsp70. It is in particular Hsp70 that has been shown to be able to induce heat resistance when over-expressed in cells Citation[32] and this could be linked to its ability to act as a chaperone in cells Citation[16], Citation[33]. Whereas over-expression of small HSPs can also give rise to expression of heat resistance Citation[34], this effect seems more cell system dependent and has not yet been clearly linked to its chaperone activity, although effects such as stabilising cytoskeletal proteins and/or accelerating post-heat recovery of heat damage have been attributed to increased Hsp27 expression.
Most HSPs, except the small HSPs, are ATP-dependent chaperones and have several co-factors that regulate their cycling between ATP- or ADP-bound states as well the binding and release of their client proteins (). For Hsp70, there are both positive (e.g. Hsp40, Hip) and negative (e.g. Bag-1) regulators of refolding activity that were not only clearly characterised in cell-free systems Citation[35–37], but their effect on the ‘Hsp70 chaperone machine’ have also been validated in living cellsCitation[15], Citation[38], Citation[39]. Since this chaperone machine has not only been implicated in the heat shock response but is linked to many pathologies, screens for drugs capable of either enhancing or reducing Hsp70 or its function as a chaperone machine are ongoing in many laboratories. Whether or not targeting each of these regulators or Hsp70 itself will be of use in hyperthermic oncology remains unclear but may not be without any risk of side-effects. Whereas constitutive Hsp70 is expressed in most normal tissues, the inducible isoform is not. On the other hand, the inducible form is expressed in many tumours in situ Citation[40]. It has been suggested that this is related to the putative role that Hsp70 may play in the apoptotic programm Citation[41], Citation[42] but this is still very controversial. It is also not clear if and how this may be related to the Hsp70 chaperone activity or maybe other activities such as lysosomal stabilisation Citation[43]. Irrespective of the molecular mechanism, drugs that would specifically target the inducible Hsp70 may be worthy of further study especially combined with hyperthermia as they would not only lead to tumour-specific toxicity by themselves but also may act as tumour-specific thermosensitisers.
Damage to structures and its consequences
Despite the fact that we now know that protein damage plays a central role in the biological effects of hyperthermia, little is known about what finally kills the cells. Nuclear proteins appear most sensitive Citation[11] and/or the nuclear environment is favourable for protein aggregation to occur Citation[44]. Aggregation of nuclear proteins has been observed since the early days of hyperthermia research Citation[45], Citation[46] and has been linked to inhibition of transcription and DNA replication Citation[12]. It can be perceived that if nuclear structures and functions are irreversibly damaged, this will result in cell death Citation[18]. Damage to the nucleus has also been closely linked to impaired processing of DNA damage Citation[47] and thus has consequences for both the response of cells to radiation and DNA targeting cytotoxic drugs. As such nuclear protein aggregation seems to play a major role in heat radiosensitisation and may also contribute to thermal sensitisation to certain chemotherapeutics.
Irreversible protein damage to non-nuclear compartments, if properly disposed by the cells (e.g. via degrading the aggregated proteins), may be replaced by new synthesis and thus less harmful. Nevertheless, damage to other compartments and/or processes also could contribute to cell death after heat shock. In this respect, especially, centrosomes should be mentioned. These structures, that are crucial in organising the mitotic spindle, are replicated once per cell cycle and need to move to the opposite poles at the onset of mitosis. It was demonstrated by Vidair et al. Citation[48] that centrosomes are highly sensitive to heat shock and recent evidence suggests Citation[49] that heat damage to centrosomes may be linked to mitotic catastrophe after hyperthermia and thus to loss of reproductive cell capacity ().
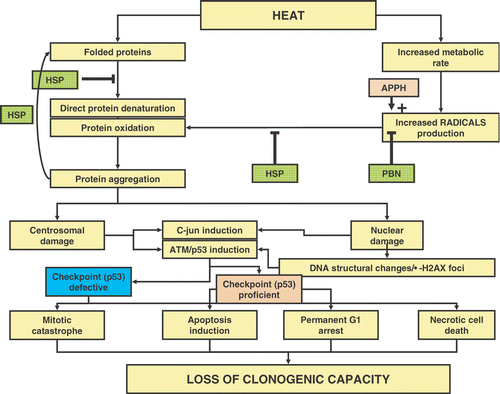
Figure 3. Hypothetical model for cell death expression after hyperthermia. See text for explanation of the details.
Cell death expression
From the consensus that protein damage plays a central role in the biological effects of heat, one can derive a global model on how this may lead to cell death (). If tumour cell death is considered as the most relevant endpoint in hyperthermic oncology (certainly normal tissue cell death cannot be neglected), the ultimate endpoint to consider is reproductive cell death. Depending on the default programme in cells, loss of reproductive ability may be the result of the induction of apoptosis or necrosis. In addition, permanent G1 arrest and mitotic catastrophe (followed by secondary apoptotic or necrotic death) will stop cells from reproduction. Protein damage (either from direct denaturation or via increased radical production leading to protein oxidation) will cause irreversible damage to nuclear (matrix) structures, maybe reflected by induction of γ-H2AX foci, or will cause irreversible non-nuclear damage to structures like the centrosome. This will induce stress signalling pathways like the JNK-pathway and the ATM/ATR pathway. In checkpoint proficient cells, p53 dependent (or independent) signals may provoke either apoptosis or a permanent G1-arrest, both leading to loss of clonogenicity. In checkpoint deficient cells, cells may either die in interphase due to necrosis (in case of irreversible nuclear damage) or progress through the cell cycle despite the irreversible damage that was inflicted to, for example, the centrosomes. At mitosis, damage to the centrosomes will result in mitotic catastrophe and most cells will die consequently, albeit with the risk that some cells may become aneuploid and hence more genetically unstable.
The scheme in implies that the mode of cell death programmes that are present (by default) in cells may be less important for the ultimate effects of heat on clonogenicity than the actual damage inflicted. Cells that are defined as heat sensitive by the clonogenic assay may not undergo apoptosis after heating if, for example, their p53 is mutated. Rather, they may die via one of the other routes. This agrees with the notion that there is no consistent intrinsic difference in reproductive heat sensitivity between normal (checkpoint and apoptosis proficient) and tumour cells (often checkpoint and apoptosis deficient), although in some cases for example the p53 status may alter the heat response of cells Citation[50]. No consensus was reached on whether the latter is really due to defective checkpoints and apoptotic programmes or due to secondary effects of the altered p53 status (affecting the handling of protein damage) and this therefore remains to be elucidated.
The scheme in is also consistent with the thermoprotective effects of HSPs; these can either prevent protein denaturation or/and assist in repair or protein aggregates. The HSPs may also protect against the protein oxidation that may be induced by heat shock consistent with their protective action against oxidative stresses Citation[26]. Finally, this scheme is also consistent with the observation that enhancement or reduction of radical production can alter heat toxicity Citation[51].
The implication of the above for hyperthermic oncology is that hyperthermia should be applicable to all tumours irrespective of their checkpoint or p53 status, although no actual systematic analysis has been done to substantiate this idea. It also re-emphasises the fact that selective anti-tumour effects of hyperthermia may not be the result of systematic differences in tumour or normal cellular response. Thus, heat targeting to tumours and hopefully the beneficial effects from physiological parameters are items that should give hyperthermia its therapeutic gain. Finally, it supports the earlier mentioned notion that modulating the ability of cells to deal with protein damage (e.g. heat shock proteins, pro- or anti-oxidant agents) indeed may form the primary target for modulating the hyperthermic sensitivity of cells.
Heat and radiation interactions
Protein damage is the central event after hyperthermia. For heat-induced radiosensitisation it is thought that nuclear protein damage is the key event. Whereas this is the consensus for hyperthermic temperatures at and above 43°C, this is less clear for effects of mild hyperthermia on radiosensitivity. Hyperthermia enhances the effectiveness of radiation, but radiation does not seem to affect the response of cells to hyperthermia. In other words, hyperthermia augments the amount of DNA that remains unrepaired, but radiation does not enhance the amount of heat-induced irreparable protein damage. Evidence for this notion was first demonstrated by the pioneer studies of Dewey and colleagues who found that hyperthermia enhances the amount of radiation-induced chromosomal aberrations without inducing chromosomal aberration by itself Citation[52]. Together this leads to a general consensus that this heat-induced enhancement of chromosomal aberrations is inhibition of repair of radiation-induced DNA damage Citation[11], Citation[47]. The inhibition is mostly due to structural effects at the level of the (higher order) chromatin organisation, maybe as reflected by the induction of γ-H2AX foci after heat shock that block the repair of certain DNA lesions (see below). As a result, more lethal DNA lesions remain. As such, heat acts as a radiation dose modifier. Genetic approaches have revealed that the presence of the two DNA DSB repair pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR), is not a pre-requisite for heat radiosensitisatio Citation[47], Citation[53]. This implies that heat-induced nuclear protein aggregation may not primarily act via these pathways, although minor effect via these routes may not be completely excluded. By exclusion and supported by some indirect biochemical evidenc Citation[47], Citation[53], heat may exert its major effects on radiosensitivity via inhibiting the repolymerisation step in base excision repair (BER) ().
Figure 4. Hypothetical model for the mechanism of heat-induced radiosensitization and the putative repair pathways involved.
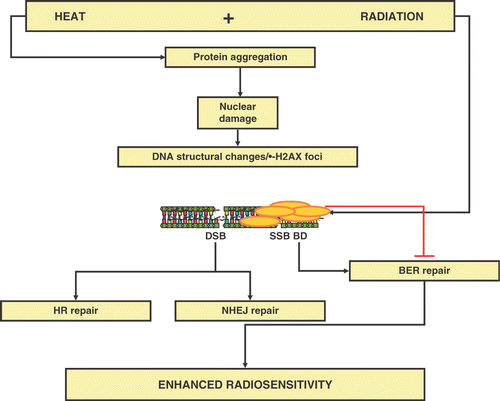
The importance of thermal radiosensitisation for the clinical application of hyperthermia is difficult to estimate. Certainly, when only short heat treatments are used (up to 1 hour), rather high temperatures (>43°C) are required to induce substantial radiosensitisation. Also, one would most benefit from the radiosensitising effects of hyperthermia if heat would be applied before or during radiation rather than a few hours after radiation as it is the current clinical practice in some medical centres. Yet, unlike previously assumed on the basis of rodent data, substantial radiosensitisation has been observed to occur even if heating is applied up to 4 hours after radiation in human cells Citation[47]. Therefore, the contribution of heat-induced radiosensitisation to the recent clinical success stories of hyperthermia cannot be excluded. In any case, the suggestion that most of the radiosensitisation may be due to effects on BER has two implications. Because deficiencies in BER are usually incompatible with cell survival, a) radiosensitisation will likely occur in all tumour types, and b) the extent of radiosensitisation most likely will not differ between normal and tumour cells. Again, this implies that therapeutic gain will have to come from selective heating devices, physiology, and maybe immunology.
Heat and drug interactions
With regard to the drug sensitising action of heat, relatively few new insights were obtained over the past decade. It still is the consensus that thermal drug sensitisation for alkylating agents like platinum-based drugs is the strongest and in theory holds great promise. Combination of heat and platinum is more than additive, and a therapeutic gain may come from the fact that cells in poorly vascularised tumour areas (that are drug resistant due to limited drug delivery) are especially sensitive to heat. Moreover, by increasing (tumour) blood flow, hyperthermia will enhance drug delivery in these poorly vascularised areas. Finally, synergy may arise from cell biological effects of heat that enhance drug accumulation and reduced intracellular detoxification and repair of platinum-induced adducts. Again, protein damage by heat may underlay these effects but the evidence for this is not yet clear. Also, cells that have acquired platinum resistance (e.g. during the course of a platinum-based chemotherapy protocol) can be made responsive to platinum again by heat. However, only in certain cases resistance was found to be completely reversed. Agents that enhance the effect of heat have been around for several decades (procaine, ethanol) but so far no clinically applicable drugs have been found that can potentiate the cellular effects of hyperthermia.
Part II. B. Genetic, immunological and physiological aspects of hyperthermia
CHANG W. SONG1, HEON J. PARK2, GLORIA LI3 & ELIZABETH A. REPASKY4
1University of Minnesota, Minneapolis, USA, 2Inha University, Inchon, Korea, 3Memoral Sloan Kettering Cancer Center, New York, USA and 4Roswell Park Cancer Institute, Buffalo, NY, USA
Hyperthermia regulated gene therapy
Altering the genetic make-up of cancer cells by gene transfer, i.e. gene therapy, is a potentially powerful strategy for treating human cancer. However, relatively poor tumour specificity and low efficiency of gene delivery in vivo have prevented the widespread implementation of this technology in the clinic. The feasibility of utilising heat shock to enhance the efficacy of gene therapy has been explored by investigators Citation[1]. For example, the effect of hyperthermia on the anti-tumour effect of an adenoviral vector coding for IL-12 placed under the control of a heat inducible promoter, i.e. promoter of the Hsp70, was investigated. Injecting the construct into murine tumours and heating the tumours 24 h later at 42°C for 40 min caused a significant increase in IL-12 level in tumours and suppressed the tumour growth Citation[2–5]. Transfection of cancer cells with plasmid (pHSp53–121F) containing mutated p53 gene (p53–121F), an inducer of apoptosis, linked to the HSP70B promoter, and heating the cells at 43°C for 2 h caused apoptosis 70-fold greater than that caused by heating alone Citation[6]. Hyperthermia regulated gene therapy may also be used to enhance the response of tumours to radiotherapy or chemotherapy Citation[7–10]. DNA-dependent protein kinase (DNA-PK) plays a central role in the repair of radiation-induced DNA double-strand break (DSB). DNA-PK consists of a large catalytic subunit and a DNA-targeting component Ku, which itself is a heterodimer of Ku-70 and Ku-86. Therefore, reduction of the cellular level of Ku may reduce the ability of DNA-PK to repair radiation-induced DSB, thereby increasing the radiosensitivity of cells. When cancer cells were infected with adenovirus vectors containing antisense Ku70 RNA under the control of an inducible hsp70 promoter and heated 24 h later, the endogenous Ku70 was markedly reduced and, consequently, the cells became sensitive to radiation both in vitro and in vivo Citation[10]. Certain anti-cancer drugs such as mitomycin C and β-lapachone are bioactivated by NAD(P) H:quinone oxidoreductase (NQO1), a two electron reductase. It has been shown that the NQO1 gene is activated by heat shock and thus the enzymatic activity of NQO1 is significantly increased demonstrating that heat may sensitise cells to the aforementioned NQO1-dependent bioreductive drugs Citation[11]. Indeed, cancer cells heated at 41°–42°C for 1 h were markedly sensitive to β-lapachone due to the heat-induced upregulation of NQO1 as long as 24–48 h after heating Citation[11]. These observations clearly indicate that heat shock is a potent means to activate transfected or endogenous genes and it may be possible to exploit such a heat-induced activation of genes to sensitise cancer cells to radiotherapy or chemotherapy. Furthermore, it may be also feasible to use hyperthermia to increase the efficacy of gene therapy to treat human ailments other than cancer. For example, mild local hyperthermia of liver regions of patients with chronic hepatitis C increased the expression of IFN-alpha receptor1 suggesting that hyperthermia may enhance the anti-viral efficacy of IFN by upregulating IFNR1 expression Citation[12].
Hyperthermia regulated tumour immunity and immunotherapy
It has become increasingly evident recently that hyperthermia is a powerful stimulator of immunological elements Citation[13–15]. It is well known that heat shock proteins (HSPs) are synthesised and released to extracellular milieus when cells are subjected to various stresses including heating. HSPs play a critical role in maintaining cell homeostasis and the development of thermotolerance. Among various heat shock proteins, Hsp70 family members have been demonstrated to play prominent role in immune response to cancer by stimulating both the innate and adaptive immune response Citation[14–19]. Specifically, Hsp70, a molecular chaperone, binds immunogenic peptides and presents them to dendritic cells (DCs), thereby eliciting antigen-specific cytotoxic T cells. In addition to functioning as a carrier of antigenic peptides, Hsp70 binds to DCs, thereby inducing DC maturation and secretion of pro-inflammatory cytokines. In this respect, heating hepatic cancers at mild temperatures induced CD16-positive immature monocytes in the circulation and activated T cells resulting in production of large amounts of type-1 cytokines Citation[20]. It would be reasonable to attribute such an increase in immunogenic activity in human patients receiving regional hyperthermia, at least in part, to a heat-induced increase in HSP production.
A relevant and promising development in recent years is the suggestion that the conjugates of HSPs with antigenic peptides may be used as an anti-cancer vacci Citation[15], Citation[16]. The underlying rationale of this approach is that HSPs function as chaperones for the antigenic proteins and present the antigens to immune systems. The choice and source of HSPs and peptides may affect the specificity and potency of the HSP-peptide-based vaccine. Further investigation is warranted to develop the potentially powerful HSP-based immunotherapy for the treatment of human cancers.
Fever-range whole body hyperthermia
Whole body hyperthermia at fever-range temperatures, e.g. 39°–40°C, (FR-WBH) has been demonstrated to enhance various immune reactions, in addition to augmenting the effect of chemotherapy Citation[13], Citation[21]. The mechanism underlying the enhancement of immunogenicity by FR-WBH has not been fully elucidated. Two mechanisms have been suggested. The first mechanism is heat-induced upregulation of HSPs, which is known to elicit an immune response as alluded to above. When rodents were exposed to FR-WBH, the expression of Hsp70 and Hsp110 increased in various organs including the heart, kidney, lung, lymph nodes and thymus Citation[21]. The second proposed mechanism is an improvement of lymphocytes trafficking across vessel walls in lymphoid tissues and also an improvement of extravasation of immune effecter cells across the blood vessel walls in target tissues such as tumours Citation[22]. Importantly, such an increase in extravasation of lymphocytes by FR-WBH occurs preferentially in lymphoid tissues, tumours or inflammatory tissues relative to normal tissues. Adhesion of lymphocytes to the vessel wall mediated by adhesion molecules such as ICAM is an initial and essential process for extravasation of blood-borne lymphocytes. FR-WBH has been shown to upregulate adhesion molecules on the surface of the vessel wall, thereby promoting lymphocytes trafficking across the blood vessel wall. It is likely that the expression of ICAM-1 is intrinsically upregulated in lymphoid tissues, tumours and inflammatory tissues, and further increase by FR-WBH may augment extravasation and accumulation of lymphocytes in these tissues.
A relevant observation is that local heating of tumours at mild temperatures, like FR-WBH, also augments expression of ICAM-1 molecules on endothelial cells in tumours, thereby increasing the extravasation of immune lymphocytes. It is unclear whether the increases in ICAM-1 in tumour endothelial cells by local heating and FR-WBH are caused by the same mechanisms. It has been suggested that the increase in ICAM-1 expression in tumour endothelial cells by local heating may result, at least in part, from an increase in sheer forces due to the heat-induce increase in tumour blood flow.
Importantly, the efficiency of a vaccine made of the tumour derived HSPs was found to be elevated by FR-WBH, probably due to enhanced extravasation and intra-tumour accumulation of T cells which are primed to be immunogenic to the tumour cells by the HSPs Citation[16]. It has also been reported that FR-WBH augments the efficacy of vaccination with autologous dendritic cells in patients with solid tumours Citation[23]. An interesting and potentially important observation is that heating at mild temperatures lower than 41°C enhanced DC maturation in vitro Citation[24]. It should be noted that DCs are abundant in the skin where they develop into competent antigen presenting Langerhans cells, and from where they migrate into the lymphatic systems to prime T cells Citation[24]. It would be reasonable, therefore, to expect that FR-WBH accelerates maturation of DCs in the skin, thereby increasing the host immunity. Pre-clinical or clinical studies of the combination of FR-WBH with chemotherapy are in progress at a number of institutes Citation[24–26]. How the immunologic status changes in patients receiving FR-WBH in combination with chemotherapy remains to be studied.
Heat-induced vascular change and tumour oxygenation
It has been well demonstrated that heating rodent tumours at 42°–43°C or higher temperatures causes vascular destruction, which results in decreased blood flow and necrosis Citation[27], Citation[28]. However, it became increasingly clear that human tumour blood vessels are heat-resistant as compared with rodent tumour blood vessels and, what is more, it is rather difficult to adequately heat human tumours, particularly bulky and deep-seated tumours, to temperature high enough to cause cell death or vascular damage in the tumours. Interestingly, however, a number of clinical trails demonstrated that combined treatment of human tumours with radiotherapy and hyperthermia was significantly more effective than radiotherapy alone for the control of the tumours despite the fact that the tumour temperature could not be raised to cytotoxic levels, i.e. >42°–43°C. A series of experiments then subsequently demonstrated that heating at mild temperatures, i.e. 39°–42°C, causes a long-lasting increase in blood flow and an increase in oxygenation in rodent tumours as well as in canine tumours Citation[29–34]. Indications are that reduction of oxygen consumption due to thermal damage in cells may also be a factor for the increase in tumour oxygenation following tumour heating Citation[34]. Subsequent studies clearly demonstrated that hyperthermia increases oxygenation also in human tumours and enhances the response of the tumours to radiotherapy Citation[35–37]. It is important to realise that temperature distribution in human tumours during heating is rather heterogeneous; the temperature in certain areas in tumours may increase to 39°–42°C range while that in other areas in the same tumours may rise to 43°–45°C. It is therefore highly likely that while blood flow increases in certain areas, it may decrease in other areas in the same tumours. This implies that hyperthermia may cause direct cell death and vascular damage accompanied by necrotic cell death in certain areas while it improves oxygenation, thereby increasing radiosensitivity of tumour cells in other areas in the same tumours. During the last several decades, a variety of different methods have been proposed and tried to increase oxygenation in human tumour, but none of them has been proven to be clinically useful. For example, carbogen (95% O2 + 5% CO2) breathing was reported to increase the oxygenation and radiosensitivity of murine tumours, but the therapeutic gain by the application of carbogen breathing was only marginal Citation[38], Citation[39]. It was recently demonstrated that the increase in oxygenation and radiosensitivity by the combination of carabogen breathing with mild temperature hyperthermia (MTH) was significantly greater than that caused by carbogen breathiCitation[32], Citation[40], Citation[41]. It is thus highly recommended to investigate the potential usefulness of MTH combined with carbogen breathing to increase oxygenation and radiosensitivity of human tumours. The efficacy of certain chemotherapy drugs is also influenced by oxygenation status of cancer cells and thus heat-induced increase in tumour oxygenation would increase the cytotoxicity of such drugs. In addition, MTH-induced increase in blood perfusion would undoubtedly increase drug uptake in solid tumours.
Overview and conclusion
CHANG W. SONG
University of Minnesota, Minneapolis, USA
The forum started with a reception the evening of the first day of the Forum (14 June 2004). During the next three days a total of 24 clinical papers, 20 biological papers and 6 reviews were presented at plenary type sessions, where both clinicians and biologists participated. Thereafter, a general discussion session was held to further exchange participants’ opinions on various subjects presented during the previous plenary sessions. On the evening of the third day, clinicians and biologists had separate sessions to draft their conclusions and recommendations, which were then presented at the adoption and proposal session on the fourth day by representatives of each group. The preceding Part I and Part II are the summary of discussions prepared by the representatives of the clinical group and biology group, respectively. The original drafts were then reviewed by key participants in each group before submission to the International Journal of Hyperthermia.
The major subjects discussed and conclusions made at the Forum are briefly addressed in this section.
The interest in clinical hyperthermia has been slowly reviving in recent years, particularly in Japan and Europe. Unfortunately, there has been little improvement in our ability to heat human tumours, particularly deep and bulky tumours to cytotoxic temperatures. Thermometry is still a major problem in clinical hyperthermia. Although non-invasive thermometry devices are being developed by several groups of investigators, they are not yet ready for routine use at hyperthermia clinics. During the last decade, numerous randomised clinical trials for hyperthermia were conducted in Europe, Japan and North America. In these studies, tumour temperatures seldom reached target temperatures, i.e. 42°–45°C. Nevertheless, hyperthermia was often demonstrated to enhance the efficacy of radiotherapy or chemotherapy to achieve local tumour control. Based on the published and unpublished reports presented at the Forum, the participants concurred that hyperthermia is effective to enhance the efficacy of radiotherapy and chemotherapy for the control of tumours of the head and neck, breast, brain, bladder, cervix, rectum, lung, oesophagus, and melanoma, and that such improvement of local tumour control increases the overall survival rate of patients. The recent trend in whole body hyperthermia (WBH) was also discussed at the Forum, and it was agreed that WBH may be useful to enhance the effect of certain anti-cancer drugs. The Japanese participants reported that in addition to curative purposes, hyperthermia is frequently used to palliate the cancer pain, thereby improving the patient's quality of life in Japan. (A number of additional consensus and recommendations on clinical hyperthermia were made. Readers are referred to Part I. Clinical hyperthermia.)
Research on the basic biology of heat shock has been steadily progressing worldwide, and impressive new insights into the heat shock response at molecular and cellular levels have been revealed in recent years. The mechanisms of heat-induced cell death, radiosensitisation, and chemosensitisation were discussed at the Forum. Heat-induced protein damage has been known to be the main molecular event underlying the cytotoxic effect of hyperthermia at the clinically relevant temperatures, i.e. 39°–45°C. However, some participants pointed out that heat shock induces γ-H2AX foci, a hallmark of DNA double strand break (DSB), and suggested that heat may kill cells by directly causing DNA DSB. Opposite to such direct DNA DSB hypothesis, other investigators proposed that the heat-induced γ-H2AX foci formation may result from the changes in chromatin structure caused by heat-induced protein aggregation with the nuclear matrix. The importance of the role of Hsps as molecular chaperones in the response of cells to heat shock and other stresses was re-confirmed at the Forum. The participants expressed considerable interest in the mechanisms underlying the HSP gene activation, and the implication of the interactions among heat shock proteins, HSF-1 and HSPB1 in the response of cells to heat shock. The molecular signal transductions leading to apoptosis and clonogenic cell death after heat shock were also addressed. Contrary to the previous view that there is no intrinsic difference in the reproductive heat sensitivity between normal cells and tumour cells, a group of investigators from Japan presented experimental evidence that the normal cells were heat resistant as compared with tumour cells in vitro, probably due to increased levels of HSP in the normal cells at confluent stage. It has been known that heat shock sensitises cells to radiation, probably by inhibiting the repair of radiation-induced DNA damage. However, the molecular mechanism underlying the inhibition of the repair of DNA damage by heat shock is still unclear, although it was suggested that heat shock increases radiation-induced chromosome aberration thereby indirectly increasing DNA DSB. It was also suggested that heat may inhibit repair of radiation-induced DNA damage by interfering with the base excision repair (BER). With regard to the heat-induced increase in the efficacy of certain chemotherapy drugs against tumour cells, the following consensus was reached:
heat enhances the damage in target molecules caused by drugs,
heat increases the cellular uptake of drugs, and
heat increases the drug delivery to target cells by increasing blood perfusion.
Experimental evidence was presented that heating at mild temperatures, i.e. 39°–42°C, increases tumour blood flow and that such an increase in tumour blood flow improves tumour oxygenation. It was also reported that treating human tumours with conventional fractionated irradiation in combination with hyperthermia at mild temperatures markedly improves the oxygenation status in the tumours. However, the role of the heat-induced increase in tumour oxygenation in the response of human tumours to radiotherapy was questioned by some participants because in some institutions hyperthermia was routinely applied several hours after radiotherapy and yet the combination of hyperthermia and radiotherapy was significantly more effective than radiotherapy alone to achieve tumour control. It was pointed out that the improvement of tumour oxygenation by mild temperature heating lasts for 24–48 hours pointing to the possibility that hyperthermia applied several hours after radiotherapy enhanced the response of tumours to subsequently applied radiotherapy the next day. It was recommended to further investigate the implications of vascular changes caused by different temperatures in the response of human tumours to radiotherapy.
It has been increasingly evident in recent years that anti-cancer immunogenicity can be upregulated by heat. Local or regional heating of tumours at mild temperatures or whole body heating at fever range temperatures (39°–41°C) enhances immune response through increased production of immunogenic heat shock proteins (HSPs), activation of antigen presenting cells (APCs) and improved trafficking of immune cells to lymphoid organs and target tumours. The feasibility of enhancing the efficacy of gene therapy using heat shock was another topic discussed at the Forum. Heat shock is a potent means to activate transfected or endogenous genes, which are cytotoxic alone or are able to increase the sensitivity of cells to radiotherapy or to certain chemotherapy drugs.
The scope of using hyperthermia to treat human diseases has been expanding in recent years. The participants in the Forum felt that the Forum provided an excellent opportunity to exchange new information and opinions regarding the potential of hyperthermia for the treatment of cancer. The final consensus at the Forum was that concerted efforts should be made by the hyperthermia community to improve the perception of medical establishments towards the effectiveness of hyperthermia. Effort should also be made to inform the general public the value of recently evolved hyperthermia for the treatment of cancer.
Notes
Correspondence: Harm H. Kampinga, Faculty of Medical Sciences, Department of Radiation and Stress Cell Biology, Division of Cell Biology, University Medical Centre Groningen, University of Groningen, Ant. Deusinglaan 1, Groningen 9713 AV, The Netherlands. Email: [email protected]
Correspondence: Chang W. Song, PhD, Radiobiology Laboratory, MMC 494, University of Minnesota Medical School, Minneapolis, MN 55455, USA.
References
- International Concensus Meeting on Hyperthermia: Final report. Int J Hyperthermia 1990; 6: 837–877
- Jorritsma JB, Konings AW. The occurrence of DNA strand breaks after hyperthermic treatments of mammalian cells with and without radiation. Radiat Res 1984; 98: 198–208
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, Mori E, Yamakawa N, Yasumoto J, Yuki K, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004; 64: 8839–8845
- Konings AWT. Membranes as targets for hyperthermic cell killing. Cancer Res 1988; 109: 9–22
- Dewey WC. Arrhenius relationships from the molecule and cell to the clinic. Int J Hyperthermia 1994; 10: 457–483
- Lepock JR, Frey HE, Ritchie KP. Protein denaturation in intact hepatocytes and isolated cellular organelles during heat shock. J Cell Biol 1993; 122: 1267–1276
- Burgman PWJJ, Konings AWT. Heat induced protein denaturation in the particulate fraction of HeLa S3 cells: Effects of thermotolerance. J Cell Physiol 1992; 153: 88–94
- Spiro IJ, Denman DL, Dewey WC. Effect of hyperthermia on CHO DNA polymerases a and b. Radiat Res 1982; 89: 134–149
- Bensaude O, Pinto M, Dubois M-F, Nguyen VT, Morange M. Protein denaturation during heat shock and related stress. Stress Proteins 1990; 8: 89–99
- Kampinga HH. Thermotolerance in mammalian cells. Protein denaturation and aggregation, and stress proteins. J Cell Sci 1993; 104: 11–17
- Lepock JR. Role of nuclear protein denaturation and aggregation in thermal radiosensitization. Int J Hyperthermia 2004; 20: 115–130
- Roti Roti JL, Laszlo A. The effects of hyperthermia on cellular macromolecules. Thermal effects on cells and tissues, M Urano, E Douple. VSP, Utrecht 1988; 13–98
- Borrelli MJ, Stafford DM, Rausch CM, Lepock JR, Lee YJ, Corry PM. Reduction of levels of nuclear-associated protein in heated cells by cycloheximide, D2 O, and thermotolerance. Radiat Res 1992; 131: 204–213
- Lepock JR, Cheng KH, Al-Qysi H, Kruuv J. Thermotropic lipid and protein transitions in Chinese hamster lung cell membranes: Relationship to hyperthermic cell killing. Can J Biochem Cell Biol 1983; 61: 421–427
- Michels AA, Kanon B, Ohtsuka K, Konings AWT, Bensaude O, Kampinga HH. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem 1997; 272: 33283–33289
- Stege GJ, Li GC, Li L, Kampinga HH, Konings AW. On the role of hsp72 in heat-induced intranuclear protein aggregation. Int J Hyperthermia 1994; 10: 659–674
- Lepock JR, Frey HE, Heynen ML, Senisterra GA, Warters RL. The nuclear matrix is a thermolabile cellular structure. Cell Stress & Chaperones 2001; 6: 136–147
- Roti Roti JL, Kampinga HH, Malyapa RS, Wright WD, vanderWaal RP, Xu M. Nuclear matrix as a target for hyperthermic killing of cancer cells. Cell Stress & Chaperones 1998; 3: 245–255
- Kampinga HH, Turkel-Uygur N, Roti Roti JL, Konings AWT. The relationship of increased nuclear protein content induced by hyperthermia to killing of HeLa S3 cells. Radiat Res 1989; 117: 511–522
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146: 905–916
- Raaphorst GP, Vadasz JA, Azzam EI. Thermal sensitivity and radiosensitization in V79 cells after BrdUrd or IdUrd incorporation. Radiat Res 1984; 98: 167–175
- Wong RSL, Dewey WC. Molecular mechanisms for the induction of chromosomal aberrations in CHO cells heated in S phase. Environ Mol Mutagen 1993; 22: 257–263
- Ritossa F. Discovery of the heat shock response. Cell Stress & Chaperones 1996; 23: 197–98
- Tissieres A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: Relation to chromosome puffs. J Mol Biol 1974; 84: 389–398
- Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int J Hyperthermia 1995; 11: 459–488
- Morimoto RI. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes & Dev 1998; 12: 3788–3796
- Satyal SH, Chen D, Fox SG, Kramer JM, Morimoto RI. Negative regulation of the heat shock transcriptional response by HSBP1. Genes & Dev 1998; 12: 1962–1974
- Ellis RJ, Van der Vies SM. Molecular chaperones. Annu Rev Biochem 1991; 60: 321–347
- Hartl FU. Molecular chaperones in cellular protein folding. Nature 1996; 381: 571–580
- Picard D. Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci 2002; 59: 1640–1648
- Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol 2003; 15: 419–424
- Li GC, Li L, Liu Y-K, Mak JY, Chen L, Lee WMF. Thermal response of rat fibroblasts stably transfected with the human 70-kDa heat shock protein-encoding gene. Proc Natl Acad Sci USA 1991; 88: 1681–1685
- Nollen EA, Brunsting JF, Roelofsen H, Weber LA, Kampinga HH. In vivo chaperone activity of heat shock protein 70 and thermotolerance. Mol Cell Biol 1999; 19: 2069–2079
- Landry J, Chretien P, Lambert H, Hickey E, Weber LA. Heat shock resistance conferred by expression of the human hsp27 gene in rodent cells. J Cell Biol 1989; 109: 7–15
- Minami Y, Hohfeld J, Ohtsuka K, Hartl FU. Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40. J Biol Chem 1996; 271: 19617–19624
- Hohfeld J, Minami Y, Hartl FU. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell 1995; 83: 589–598
- Takayama S, Bimston DN, Matsuzawa S, Freeman BC, Aime-Sempe C, Xie Z, Morimoto RI, Reed JC. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J 1997; 16: 4887–4896
- Nollen EA, Brunsting JF, Song J, Kampinga HH, Morimoto RI. Bag1 functions in vivo as a negative regulator of Hsp70 chaperone activity. Mol Cell Biol 2000; 20: 1083–1088
- Nollen EA, Kabakov AE, Brunsting JF, Kanon B, Höhfeld J, Kampinga HH. Modulation of in vivo HSP70 chaperone activity by Hip and Bag-1. J Biol Chem 2001; 276: 4677–4682
- Jaattela M. Escaping cell death: Survival proteins in cancer. Exp Cell Res 1999; 248: 30–43
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000; 2: 469–475
- Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY. Role of Hsp70 in regulation of stress-kinase JNK: Implications in apoptosis and aging. FEBS Lett 1998; 438: 1–4
- Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004; 200: 425–435
- Michels AA, Nguyen VT, Konings AW, Kampinga HH, Bensaude O. Thermostability of a nuclear-targeted luciferase expressed in mammalian cells–Destabilizing influence of the intranuclear microenvironment. Eur J Biochem 1995; 234: 382–389
- Roti Roti JL, Winward RT. The effects of hyperthermia on the protein-to-DNA ratio of isolated HeLa cell chromatin. Radiat Res 1978; 74: 159–169
- Tomasovic SP, Turner GN, Dewey WC. Effect of hyperthermia on nonhistone proteins isolated with DNA. Radiat Res 1978; 73: 535–552
- Kampinga HH, Dikomey E. Hyperthermic radiosensitization: Mode of action and clinical relevance. Int J Radiat Biol 2001; 77: 399–408
- Vidair CA, Huang RN, Doxsey SJ. Heat shock causes protein aggregation and reduced protein solubility at the centrosome and other cytoplasmic locations. Int J Hyperthermia 1996; 12: 681–695
- Nakahata K, Miyakoda M, Suzuki K, Kodama S, Watanabe M. Heat shock induces centrosomal dysfunction, and causes non-apoptotic mitotic catastrophe in human tumour cells. Int J Hyperthermia 2002; 18: 332–343
- Matsumoto H, Takahashi A, Wang X, Ohnishi K, Ohnishi T. Transfection of p53-knockout mouse fibroblasts with wild-type p53 increases the thermosensitivity and stimulates apoptosis induced by heat stress. Int J Radiat Oncol Biol Phys 1997; 38: 1089–1095
- Li FJ, Kondo T, Zhao QL, Hayashi Y, Ogawa R, Cui ZG, Feril LB, Jr. A lipophilic free radical initiator, 2,2′-azobis (2,4-dimethylvaleronitrile) (AMVN) enhances caspase-dependent apoptosis induced by hyperthermia. Int J Hyperthermia 2003; 19: 165–177
- Dewey WC, Sapareto SA. Radiosensitization by hyperthermia occurs through an increase in chromosomal aberrations. Cancer Therapy by Hyperthermia and Radiation. Munich, Urban & Schwarzenberg 1978; 149–150
- Kampinga HH, Hiemstra YS, Konings AW, Dikomey E. Correlation between slowly repairable double-strand breaks and thermal radiosensitization in the human HeLa S3 cell line. Int J Radiat Biol 1997; 72: 293–301
- Li CY, Dewhirst MW. Hyperthermia-regulated immunogene therapy. Int J Hyperthermia 2002; 18: 586–596
- Huang Q, Hu JK, Lohr F, Zhang L, Braun R, Lanzen J, et al. Heat-induced gene expression as a novel targeted cancer gene therapy strategy. Cancer Res 2000; 60: 3435–3439
- Siddiqui F, Li CY, Larue SM, Poulson JM, Avery PR, Pruitt AF, Zhang X, Ulrich RL, Threall DE, Dewhirst MW, Hauck ML. A phase 1 trial of hyperthermia-induced interleukin-12 gene therapy in spontaneously arising feline soft tissue sarcomas. Mol Cancer Ther 2007; 6: 380–389
- Siddiqui F, Li CY, Zhang X, Larue SM, Dewhirst MW, Ulrich RL, Avery PR. Characterization of a recombinant adenovirus vector encoding heat-inducible feline interleukin-12 for use in hyperthermia-induced gene-therapy. Int J Hyperthermia 2006; 22: 117–134
- Siddiqui F, Ehrhart AEJ, Charles B, Laura C, Li CY, Zhang X, Larue SM, Avery PR, Dewhirst MW, Ulrich RL. Anti-angiogenic effects of interleukin-12 delivered by a novel hyperthermia induced gene construct. Int J Hyperthermia 2006; 22: 587–606
- Arimura M, Suzuki K, Watanabe. New hyperthermic cancer therapy using the heat- inducible p53 gene. The Kadota Fund International Forum 2004; 81
- Blackburn RV, Galoforo SS, Corry PM, Lee YJ. Adenoviral-mediated transfer of a heat-inducible double suicide gene into prostate carcinoma cells. Cancer Res 1998; 58: 1358–1362
- Borrelli MJ, Schoenherr DM, Wong A, Bernock LJ, Corry PM. Heat-activated transgene expression from adenovirus vectors infected into human prostate cancer cells. Cancer Res 2001; 61: 1113–1121
- Lohr F, Hu K, Huang Q, Zhang L, Samulski TV, Dewhirst MW, Li CY. Enhancement of radiotherapy by hyperthermia-regulated gene therapy. Int J Radiat Oncol Biol Phys 2000; 48: 1513–1518
- Li GC, He F, Shao X, Urano M, Shen L, Kim D, Borrelli M, Leibel SA, Gutin PH, Ling CC. Adenovirus-mediated heat-activated antisense ku70 expression radiosensitizes tumor cell in vitro and in vivo. Cancer Res 2003; 633: 268–3274
- Park HJ, Choi EK, Choi J, Ahn KJ, Kim EJ, Ji IM, et al. Heat-induced up-regulation of NAD(P)H:Quinone Oxidoreductase potentiates antisequence effects of B-lapachone. Clin Cancer Res 2005; 11: 8866–8871
- Ueda H, Tanaka H, Kida Y, Ichinose M, Ostapenko VV, Miyano M, Nishide I, Yukawa S. Local hyperthermia induces expression IFNAR1 in liver of patient with chronic hepatitis C. The Kadota Fund International Forum 2004; 63
- Repasky E, Issels R. Physiological consequences of hyperthermia: Heat, heat shock proteins and the immune response. Int J Hyperthermia 2002; 18: 486–489
- Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins?. Int J Hyperthermia 2005; 21: 713–716
- Wang XY, Li Y, Yang G, Subjeck JR. Current ideas about applications of heat shock proteins in vaccine design and immunotherapy. Int J Hyperthermia 2005; 21: 717–722
- Manjili MH, Wang XY, Park J, MacDonald IJ, Li Y, Van Schie CAA, Subjeck JR. Cancer immunotherapy: Stress proteins and hyperthermia. Int J Hyperthermia 2002; 18: 506–520
- Calderwood SK, Asea A. Targeting HSP70-induced thermotolerance for design of thermal sensitizers. Int J Hyperthermia 2002; 18: 597–608
- Menoret A, Chaillot D, Callahan M, Jacquin C. Hsp70, an immunological actor playing with the intracellular self under oxidative stress. Int J Hyperthermia 2002; 18: 490–505
- Milani V, Noessner E, Ghose S, Kuppner M, Ahrens B, Scharner A, Gastpar R, Issels RD. Heat shock protein 70: Role in antigen presentation and immune stimulation. Int J Hyperthermia 2002; 18: 563–575
- Miyazawa M, Kawahara S, Kida Y, Ostrapenka V. Activation of peripheral blood T lymphocytes and induction of CD16+ immature monocytes by a regional hyperthermic treatment of the upper abdomen. The Kadota Fund International Forum. 2004; Page 61
- Ostberg JR, Kaplan KC, Repasky EA. Induction of stress proteins in a panel of mouse tissues by fever-range whole body hyperthermia. Int J Hyperthermia 2002; 18: 552–562
- Hah A, Unger E, Bain MD, Bruce R, Bodkin J, Ginnetti J, Wang WC, Seon B, Stewart CC, Evan SS. Cytokine and adhesion molecule expression in primary human endothelial cells stimulated with fever-range hyperthermia. Int J Hyperthermia 2002; 18: 534–551
- Gorter RW, Pulido E, Butorac M. Fever-range, total-body hyperthermia in combination with autologous dendritic cells in cancer patients. The Kadota Fund International Forum 2004; 41
- Kosaka M, Yamane M, Oppermann S, Simon CE. Physiology and pathophysiology of hyperthermia viewed from hyperthermic oncology. The Kadota Fund International Forum 2004; 75
- Pritchard MT, Xu Y, Kraybill W, Repasky EA. The anti-tumor effects of interleukin-12 or DOXIL are each enhanced by addition of mild (fever-range) thermal therapy. The Kadota Fund International Forum 2004; 65
- Bull JMC, Scott GL, Strebel FR, Oliver DH, Raval B, Steven M. Results of a phase I clinical trial using fever-range whole-body hyperthermia (FR-WB-TT) + cisplatin (CIS) + gemcitabine (GEM) + metronomic low-dose interferon-a (IFN-œ) and description of a phase II clinical trial to treat locally advanced and inoperable pancreas cancer. The Kadota Fund International Forum 2004; 77
- Song CW. Effect of local hyperthermia on blood flow and microenvironment: A review. Cancer Res 1984; 44: 4721s–4730s
- Reinhold HS. Physiological effects of hyperthermia. Recent Results Cancer Res 1988; 107: 32–43
- Song CW, Park H, Griffin RJ. Improvement of tumor oxygenation by mild hyperthermia. Radiat Res 2001; 155: 512–528
- Shakil A, Osborn JL, Song CW. Changes in oxygenation status and blood flow in a rat tumor model by mild temperature hyperthermia. Int J Radiat Oncol Biol Phys 1999; 43: 859–865
- Iwata K, Shakil A, Hur WJ, Makepeace CM, Griffin RJ, Song CW. Tumour pO2 can be increased markedly by mild hyperthermia. Br J Cancer 1996; 74(Suppl. 27)217–221
- Song CW, Shakil A, Griffin RJ, Okajima K. Improvement of tumor oxygenation status by mild temperature hyperthermia alone or in combination with carbogen. Semin Oncol 1997; 24: 626–632
- Okajima K, Griffin RJ, Iwata K, Shakil A, Song CW. Tumor oxygenation after mild-temperature hyperthermia in combination with carbogen breathing: Dependence on heat dose and tumor type. Radiat Res 1998; 149: 294–299
- Vujaskovic Z, Poulson JM, Gaskin AA, Thrall DE, Page RL, Charles HC, MacFall JR, Brizel DM, Meyer RE, Dewhirst MW. Temperature-dependent changes in physiologic parameters of spontaneous canine soft tissue sarcomas after combined radiotherapy and hyperthermia treatment. Int J Radiat Oncol Biol Phys 2000; 46: 179–186
- Dewhirst MW. Radiation therapy and hyperthermia improve the oxygenation of human soft tissue sarcomas. Cancer Res 1996; 56: 5347–5350
- Jones EL, Prosnitz LR, Dewhirst MW, Marcom PK, Hardenbergh PH, Marks LB, Brizel DM, Vujaskovic Z. Thermochemoradiotherapy improves oxygenation in locally advanced breast cancer. Clin Cancer Res 2004; 10: 4287–4293
- Vujaskovic Z, Song CW. Physiological mechanisms underlying heat-induced radiosensitization. Int J Hyperthermia 2004; 20: 163–174
- Inch WR, McCredie JA, Sutherland RM. Effect of duration of breathing 95% oxygen plus 5% carbon dioxide before X-irradiation on cure of C3H mammary tumor. Cancer 1970; 25: 926–931
- Siemann DW, Hill RP, Bush RS. The importance of pre-irradiation breathing times of oxygen and carbogen (5% CO2, 95% O2) on the in vivo radiation response of a murine sarcoma. Int J Radiat Biol Phys 1997; 2: 903–911
- Griffin RJ, Okajima K, Barrios B, Song CW. Mild temperature hyperthermia combined with carbon breathing increases tumor partial pressure of oxygen (pO2) and radiosensitivity. Cancer Res 1996; 56: 5590–5593
- Griffin RJ, Ogawa A, Williams BW, Song CW. Hyperthermic enhancement of tumor radiosensititization strategies. Immunol Inves 2005; 34: 343–359