Abstract
Purpose: The goal of this study was to determine whether the heat-induced formation of γ-H2AX foci is involved in hyperthermic cell killing.
Materials and methods: The heat-induced γ-H2AX response was determined in cells exhibiting various degrees of heat sensitivity. The panel of cells tested included cells that are transiently thermotolerant, permanently heat resistant, permanently heat sensitive, and permanently resistant to oxidative stress. Cells exposed to non-thermal environmental conditions that lead to protection from, or sensitization to, heat were also tested. The heat sensitivity of cells in which H2AX was knocked out was also ascertained.
Results: The protein synthesis independent state of thermotolerance, but not the protein synthesis dependent state of thermotolerance, was found to be involved in the attenuation of the γ-H2AX response in thermotolerant cells. The initial magnitude of the γ-H2AX response was found to be the same in all cell lines with altered heat sensitivity. Furthermore, no differences in the resolution of γ-H2AX foci were found among the cell lines tested. We also found that H2AX knock-out cells were not more heat sensitive.
Conclusions: We conclude that the heat-induced γ-H2AX response does not play a role in heat-induced cell killing, thereby adding further evidence that the heat-induced γ-H2AX foci are not due to DNA double strand breaks.
Introduction
The rapid phosphorylation of the carboxyterminal SQE motif of histone H2AX and the appearance of this phosphorylated form in cytologically observable foci, called γ-H2AX foci, is one of the earliest events in the orchestrated cellular response to DNA double strand breaks (DSB) generated by a great variety of agents that induce genotoxic stress, including ionizing radiation Citation1, Citation2. For ionizing radiation there is a good correlation between the number of γ-H2AX foci and the number of DSB Citation3, and the formation of γ-H2AX foci has been used as a criterion for the induction of DNA DSB Citation2. Heat has been shown to induce γ-H2AX foci by several different laboratories Citation4–6. The original report interpreted such induction of γ-H2AX foci by heat as evidence for DNA DSB induced by heat and that they play a role in hyperthermic cell killing Citation4. With respect to DNA DSB induction by heat, a recent study did not find evidence for heat-induced DNA DSB Citation6. We found that while heat induces γ-H2AX foci, (60–70 γ-H2AX foci after 60 min at 43°C, a number of foci found after irradiation with 2Gy), it does not induce DNA DSB after this heat dose, as measured by three different techniques. The limit of the resolution of one of our techniques, G2 chromosomal aberrations, was ∼4–9 DNA DSB. Thus while cells heated at 43°C for 60 min displayed a number of γ-H2AX foci equivalent to that found after 2Gy, (resulting in 70–80 DNA DSB), the heated cells contain less than ∼4–9 DNA DSB.
Nevertheless heating of cells at temperatures ranging from 41–45°C induces the formation of γ-H2AX foci Citation4, Citation6. The question addressed in this study is whether or not the process that results in and/or is associated with the formation of γ-H2AX foci in heated cells is involved in hyperthermic cell killing. If the heat-induced γ-H2AX response is due to DNA DSB (albeit a low number, as outlined above), which in turn would be expected play a role in cell killing, then it would be predicted that the γ-H2AX response would be modulated in cells with different heat sensitivities. In this report we determined the initial magnitude of heat-induced γ-H2AX response and its resolution in cells rendered thermotolerant by various treatments, in several different cell lines which are resistant or sensitive to heat-induced cell killing, and in cells treated with various agents resulting in heat resistance or heat sensitivity. Finally, we also determined the heat sensitivity of H2AX knock-out cells.
Materials and methods
Cell lines and cell culture
Chinese hamster HA-1 wild type cells and the HR-1 heat resistant variant cells, and OC–14 oxidative stress resistant variant isolated from them, were grown in minimal essential medium (MEM) supplemented with 10% fetal calf serum, non-essential amino acids and antibiotics. O23 Chinese hamster lung cells and two cell lines, 1–2 and 2–2, transfected with human hsp27 were grown in Dulbecco's modified Eagle's medium (D-MEM) supplemented with 10% fetal calf serum and antibiotics. CHO-10B2 wild type cells and HS-36, a permanently heat sensitive variant derived from them, were grown in Hams F-12 medium supplemented with 10% fetal calf serum and antibiotics. Wild type and H2AX−/− MEFs (gifts of A. Nussenzweig) were grown in DME supplemented with 10% fetal calf serum and sodium pyruvate. All cell lines were grown at 37°C, in a humidified atmosphere and 5% CO2.
For immunofluorescence studies, cells were grown on 9 × 9 mm coverslips (BellCo Glass, NJ), which have been pretreated with a few drops of glacial acetic acid in 95% ethanol, then rinsed thoroughly in distilled water and stored in 70% ethanol.
Heating
Before heating, 60 mm dishes containing coverslips were sealed with parafilm, incubated in a 37°C water-bath for 10 min followed by immersion into a precision controlled water-bath at 43°C (±0.05°C), for appropriate periods of time. Coverslips were fixed immediately or at various times post treatment, and then prepared for immunofluorescence.
Immunofluorescence
Immunofluorescence was performed as described Citation7. Briefly, cells were fixed in PBS containing 3.7% formaldehyde and 0.2% TritonX-100 for 10 min at room temperature, washed three times with phosphate buffered saline (PBS), treated with acetone at −20°C for 10 min and washed three times with PBS. The polyclonal and monoclonal γ-H2AX antibodies were obtained from Upstate Biotechnology, while Alexa-595 tagged secondary antibodies were obtained from Molecular Probes. Cells with greater than 7 or 8 γ-H2AX foci depending on the cell line, and the same brightness were counted as being positive, a method used by other workers Citation8, Citation9.
Clonogenic survival
Clonogenic survival was determined as described Citation10. H2AX+/+ and H2AX−/− MEFs were heated at 43°C for various lengths of time, or irradiated with different doses of 250 Kev X-rays, trypsinized, and plated at appropriate dilutions. Colonies were allowed to grow for 10 days, fixed, stained with methylene blue and the colonies were counted.
Treatment with amino acid analogs
HA-1 cells were treated with 2.5 mM azetidine-carboxylic acid, an analog of proline for 8 hours in MEM lacking non-essential amino acids, supplemented with 10% dialyzed fetal calf serum. Treatment with 1.0 mM canavanine, an analog of arginine for 8 hours was performed in arginine-free MEM, supplemented with 10% dialyzed fetal calf serum.
Glycerol treatment
HA-1 cells were incubated with 1 M glycerol in complete medium for 30 min at 37°C. Following such incubation the medium was changed and the cells were immediately heated at 43°C for various lengths of time.
Results
Which state of thermotolerance is involved in the attenuation of the γ-H2AX response?
It has been reported that the magnitude of γ-H2AX response is attenuated in thermotolerant cells Citation4. The following sets of experiments were designed to determine which two components of thermotolerance, the protein synthesis dependent (PSD) and protein synthesis independent (PSI) Citation11, Citation12 affect the γ-H2AX response and if recovery from the response is enhanced in thermotolerant cells. The initial report about the attenuation of the γ-H2AX response in thermotolerant (TT) cells did not determine which of the two states of thermotolerance, Citation11, Citation12, may be involved, and also did not examine the decay of such attenuation nor the resolution of the response Citation4. One of the hallmarks of thermotolerant cells is the more rapid recovery from cellular perturbations involved in heat-killing Citation13, Citation14.
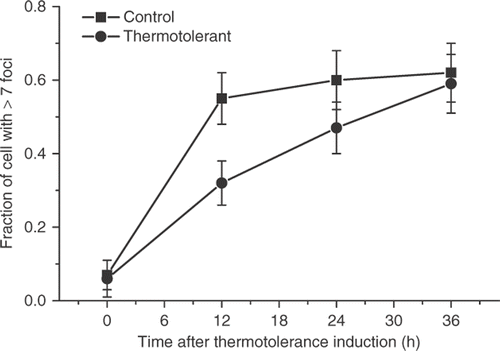
When thermotolerance in HA-1 cells was induced by 30 min at 43°C treatment, followed by recovery for 18 hours at 37°C, the induction of γ-H2AX foci by increasing doses at 43°C was attenuated when compared to control cells (). (The typical induction of foci containing γ-H2AX by 60 min heating of control HA-1 cells at 43°C is illustrated in ). This observation confirms those reported earlier Citation4. Determination of the decay of the attenuated γ-H2AX response in heat-induced TT cells indicated that the effect was significantly diminished at 24 hours and not observed at 36 hours after the induction of thermotolerance ().
Figure 1. The effect of thermotolerance on the initial magnitude of the heat-induced γ-H2AX response. Untreated HA-1 cells (▪), HA-1 cells heated at 43°C for 30 min and allowed to recover at 37°C for 12 h (•) and HA-1 cells treated with 100 μM sodium arsenite for 1 h and allowed recover at 37°C for 6 h (▴) were heated for increasing times at 43°C, processed for immediately for immunofluorescence, and the number of γ-H2AX positive cells determined as described under Materials and methods.
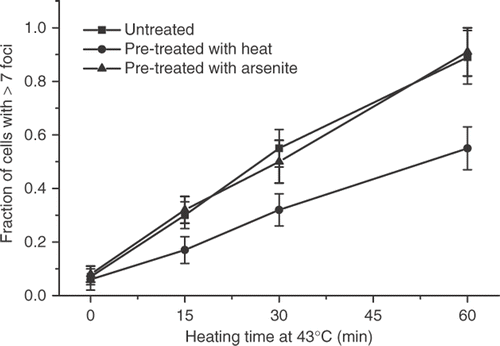
Figure 2. Heat induces the formation of γ-H2AX-containing foci in HA-1 cells. Untreated cells (A) and cells heated at 43°C for 60 min (B) were fixed and stained with an antibody against γ-H2AX described under Materials and methods.
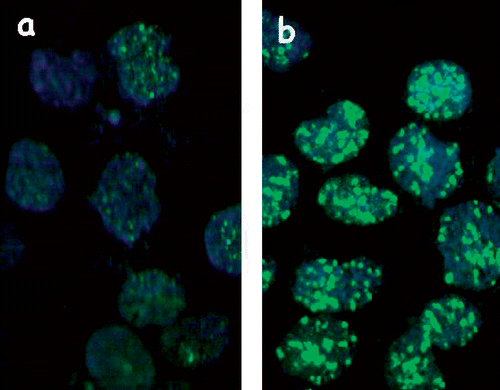
Figure 3. The decay of the attenuation of the γ-H2AX response in thermotolerant HA-1 cells. Control cells (▪) and cells heated at 43°C for 30 min allowed to recover for 12 h (•) were heated at 43°C for 60 min at various times post treatment and the fraction of cells positive for γ-H2AX foci cells was determined.
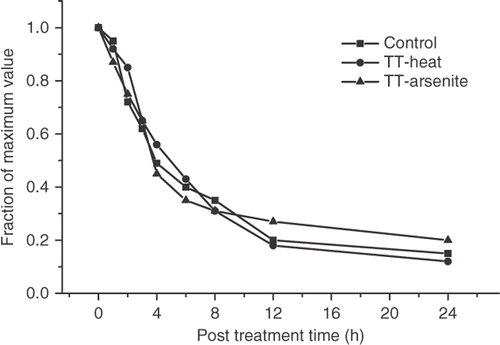
This result is in contrast with clonogenic thermotolerance, which decays between 72–96 hours after its induction under these experimental conditions Citation7. To determine if the PSD or PSI thermotolerance was involved in the attenuation of the γ-H2AX response in thermotolerant cells, thermotolerance was induced by a treatment with 100 μM of sodium arsenite for 1 h followed by recovery for 12 h, a treatment that has been shown to induce thermotolerance in HA-1 cells Citation13. No attenuation of the γ-H2AX response was observed under these experimental conditions (). The fact that thermotolerance for γ-H2AX induction was not induced by sodium arsenite and that the attenuation of the γ-H2AX response decayed faster than clonogenic thermotolerance, suggested that the PSI state of thermotolerance Citation11, Citation12, may be involved in the attenuation of the γ-H2AX response in heat-induced TT cells. HA-1 cells were heated for 30 min at 43°C and allowed to recover in the presence of 100 μg/ml of cycloheximide for 6 hours. The attenuation of the heat-induced γ-H2AX response was found in such cells ().
Figure 4. Attenuation of the γ-H2AX response in cells recovering from a thermotolerance induction protocol in the absence or presence of cycloheximide in HA-1 cells. The fraction of cells positive for γ-H2AX foci was determined in control cells (▪), cells heated at 43°C for 30 min and allowed to recover for 12 h (•) and cells recovering from a 43°C heating for 30 min in the presence of 100 μM cycloheximide for 6 h (▴).
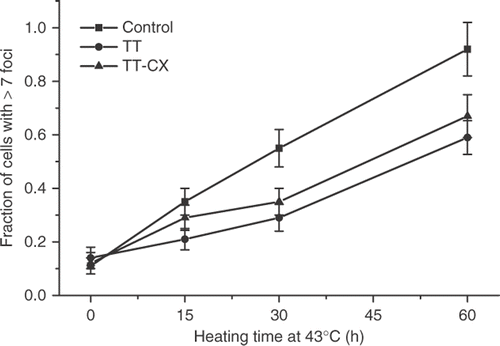
Thus, the attenuation of the γ-H2AX response is associated with the PSI state of thermotolerance. Comparison of the resolution of the γ-H2AX foci in control and thermotolerant cells () indicated that this process was not faster in thermotolerant than in control cells.
Figure 5. Resolution of γ-H2AX foci in control and thermotolerant HA-1 cells. Control (▪) heat-induced thermotolerant (•) and arsenite-induced thermotolerant (▴) HA-1 cells were heated at 43°C for 60 min at various times after the original thermotolerance inducing treatment and the fraction of γ-H2AX foci positive cells was determined.
The role of molecular chaperones in the attenuation of the heat-induced γ-H2AX response
Thermotolerance induced by different agents is known to be associated with the increased expression of molecular chaperones Citation15, Citation16. The possibility that molecular chaperones play a role in the attenuation of the γ-H2AX response was ascertained by determining the heat-induced γ-H2AX response in cell lines that are permanently heat resistant due to the overexpression of molecular chaperones. HR-1 cells are a heat-resistant variant of HA-1 hamster cells that overexpresses hsc70 due to gene amplification Citation17. Clone 1.2 and 2.2 are two heat-resistant cell lines derived from O23 Chinese hamster cells in which the human hsp27 is overexpressed Citation18. There is an increased expression of hsc70 and hsp27 in thermotolerant cells Citation18–20. A more rapid recovery from heat-induced perturbation of cellular physiology associated with hyperthermic cell killing was reported in these two types of permanently heat-resistant cell lines Citation13,Citation21. We found that neither the initial magnitude of the heat-induced γ-H2AX response after heating at 43°C for various lengths of time, nor its resolution was altered in the two different permanently heat-resistant cell lines when compared to their wild type counterparts ( and ).
Figure 6. Comparison of the heat-induced γ-H2AX response in wild type HA-1 cells and the HR-1 permanently heat resistant cells derived from them. (A) HA-1 (▪) and HR-1 cells (•) were heated for various lengths of time at 43°C and the fraction of γ-H2AX foci positive cells was determined. (B) Resolution of the heat-induced γ-H2AX foci in HA-1 (▪) and HR-1 cells (•). Cells were heated at 43°C for 60 min and the fraction of γ-H2AX foci positive cells was determined at various times post treatment.
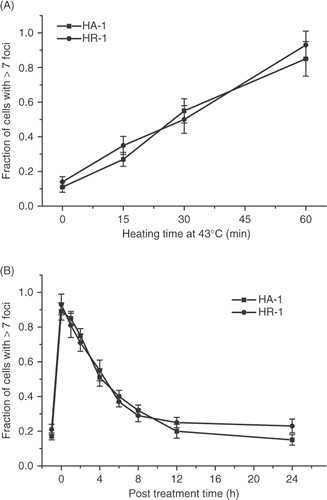
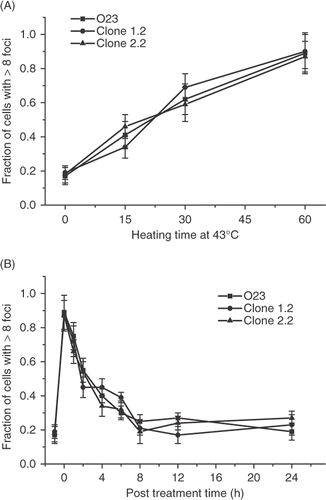
Figure 7. Comparison of the heat-induced γ-H2AX response in wild type O23 cells and two heat resistant cell lines derived from them that overexpress human hsp 27. (A) Control O23 cells (▪) and heat resistant clones 1.2 (•) and 2.2 (▴) were all heated at 43°C for various lengths of time and the fraction of γ-H2AX foci positive cells was determined. (B) Control O23 cells (▪) and clones 1.2 (•) and 2.2 (▴) were heated at 43°C for 60 min and the fraction of cells positive for γ-H2AX foci was determined at various times post treatment.
Permanently heat-sensitive cells
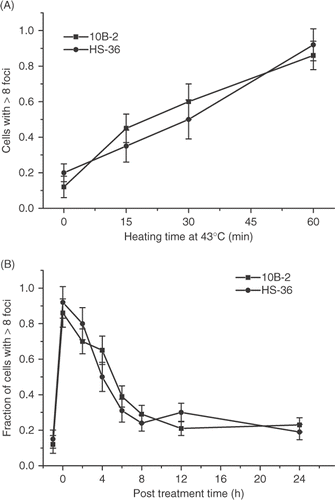
HS-36 is a permanently heat-sensitive cell line derived from Chinese hamster CHO-10B2 cells Citation22. The mechanism of the heat sensitivity has not been elucidated. However, although the magnitude of initial perturbations in cellular physiology associated with hyperthermic cell killing was similar in HS-36 cells when compared with its wild type counterpart, the recovery from such perturbations is delayed Citation21. The initial magnitude of the γ-H2AX response induced by heating for different times at 43°C was similar in HS-36 and wild type cells (). The time course of the resolution of the heat-induced γ-H2AX foci was found not to differ in the two cell types ().
Figure 8. Comparison of the heat-induced γ-H2AX response in wild type 10B-2 CHO cells and the heat sensitive HS-36 cell line derived from them. (A) Wild type 10B-2 (▪) and heat sensitive HS-36 (•) cells were heated at 43°C for various lengths of time and the fraction of γ-H2AX positive cells was determined. (B) 10B-2 cells (▪) and HS-36 cells (•) were heated at 43°C for 60 min and the fraction of γ-H2AX foci positive cells was determined at various times post treatment.
Transiently heat-resistant and heat-sensitive cells
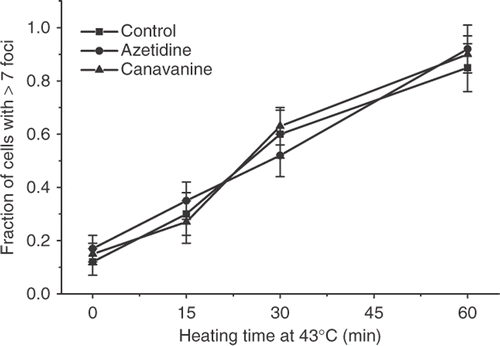
Treatment of cells with 1 M glycerol prior to exposure to elevated temperatures has been shown to protect them from heat-induced cell killing Citation23. This treatment has also been shown to protect cells from protein denaturation and aggregation Citation24–26. The heat-induced γ-H2AX response was compared in control and glycerol treated cells. Although there might be a slight protection after 15 min at 43°C, no protection was observed at either 30 min or 60 min at 43°C (). Treatment with the amino acid analog of arginine, canavanine or the proline analog azetidine has been shown to sensitize cells to heat-induced cell killing by two orders of magnitude and to inhibit the development of thermotolerance Citation27, Citation28. HA-1 cells were treated with canavanine or azetidine for 8 hours, and the induction of γ-H2AX foci after heating at 43°C for various lengths of time was determined (). There was no significant difference between the response of the control and amino acid analog treated cells.
Figure 9. The effect of glycerol on the heat-induced γ-H2AX response in HA-1 cells. Control cells (▪) and cell treated with 1 M glycerol for 30 min (•) were heated at 43°C for various lengths of time and the fraction of cells positive for γ-H2AX foci was determined.
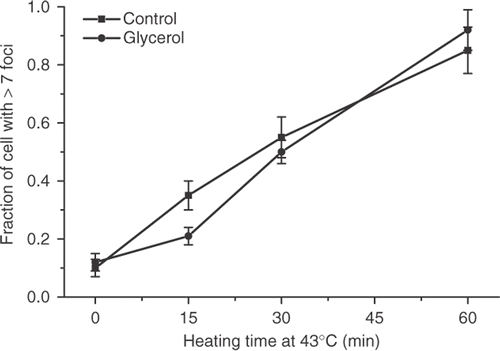
Figure 10. The effect of treatment with amino acid analogs on the heat-induced γ-H2AX response in HA-1 cells. Control HA-1 cells (▪), and HA-1 cells treated for 8 hours with 2.5 mM azetidine (•) or HA-1 cells treated for 8 hours with 1 mM canavanine (▴) were exposed to 43°C for various lengths of time and the fraction of cells positive for γ-H2AX foci was determined.
Oxidative stress resistant cells
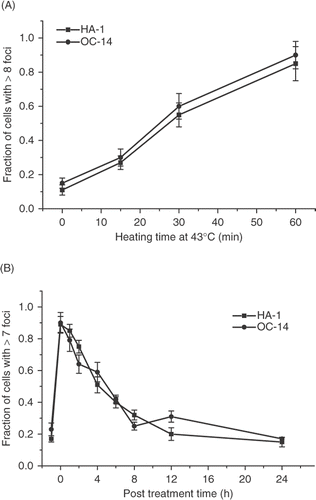
It has been suggested that the heat-induced γ-H2AX foci are not due to DNA DSB induced by heat directly, but are rather associated with the oxidative stress associated with the heat treatment Citation29. Such oxidative stress is proposed to interfere with the excision step of the nucleotide excision repair of heat-induced DNA base damage, leading to DNA DSB Citation29. OC-14 cells were selected from HA-1 cells for resistance to H2O2 Citation30. This cell line is also resistant to other oxidative stress-inducing agents, such as hyperoxia and 4-hydroxy-2-nonenal Citation31. The resistance to oxidative stress of OC-14 cells has been associated with elevated levels of catalase and glutathione Citation30, Citation32. OC-14 cells have also been reported to display resistance to heat Citation33. The initial magnitude of the γ-H2AX response after treatment at 43°C for various lengths of time was not different in OC-14 cells and wild type HA-1 cells (). The resolution of the γ-H2AX foci was also not different in the two cell types ().
Figure 11. Comparison of the γ-H2AX response of wild type HA-1 cell and the OC-14 oxidative stress resistant cells derived from them. (A) HA-1 cells (▪) and OC-14 cells (•) were heated at 43°C for various lengths of time and the fraction of positive cells for γ-H2AX foci was determined. (B) HA-1 (▪) and OC-14 (•) cells were heated at 43°C for 60 min and the fraction of cells positive for γ-H2AX foci was determined at various times post treatment.
Lack of correlation between heat sensitivity and the heat-induced γ-H2AX response
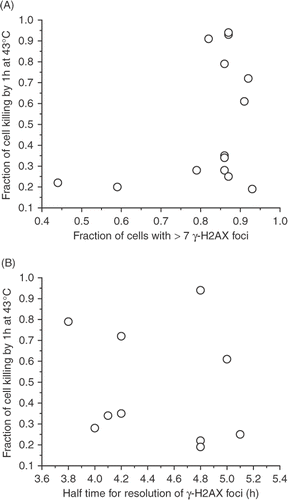
summarizes the characterization of the initial magnitude and the resolution of the heat-induced formation of foci containing γ-H2AX, and heat-induced cell killing associated with a 60-min treatment at 43°C. The heat-induced cell killing is not correlated with either the initial fraction of cells that contain >7 γ-H2AX foci, nor the half time of the resolution of the response (). Furthermore, the overall decay curve of the response is similar in all the wild type/control and differing heat sensitivity pairwise combinations that were examined (). These results constitute strong evidence that heat-induced cell killing and the heat-induced γ-H2AX response are not related.
Figure 12. Lack of correlation between heat-induced cell killing after heating for 60 min at 43°C and the fraction of cells with >7 γ-H2AX foci immediately after heating (A) or the half-time for the resolution of the γ-H2AX foci, as indicated by the decrease in the fraction of cells with >7 γ-H2AX foci to 50% of the original value.
H2AX knock-out cells
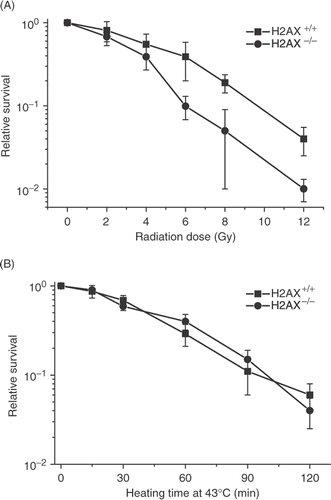
We also ascertained the heat sensitivity of cells in which H2AX has been knocked out, as a further test of whether or not the γ-H2AX response plays a role in the heat-induced cell killing. Although the H2AX knock out cells were radiosensitive, as reported before Citation34, Citation35, they did not display greater heat sensitivity or resistance ().
Figure 13. Comparison of the radiation and heat response of H2AX+/+ and H2AX−/− MEFs. (A) H2AX+/+ (▪) and H2AX−/− MEFs (•) were irradiated with various doses of X-rays and clonogenic survival was determined. (B) H2AX+/+ (▪) and H2AX−/−(•) MEFs were heated at 43°C for various lengths of time and clonogenic survival was determined.
Discussion
Heat induces the formation of γ-H2AX foci. One laboratory reported that heat induces DNA DSB by using the neutral comet assay Citation4. A recent report indicates that heat does not induce DNA DSB, using pulse field electrophoresis, the fluorescent halo assay and G2 type chromosomal aberrations Citation6. To further determine if heat-induced γ-H2AX foci were related to DNA DSB, we assayed how the induction of γ-H2AX foci by heat is associated with heat-induced cell killing. The observations that there is a dose response for the induction of γ-H2AX foci and there is thermotolerance for the effect have been used to support this idea Citation4. We reasoned if the induction and/or resolution of the γ-H2AX foci play a role in heat-induced cell killing, then this response may be modified in cells in which hyperthermic cell killing has been modified. We tested this hypothesis by determining the γ-H2AX response in cells that are resistant or sensitive to hyperthermic cell killing, including thermotolerant cells, a panel of heat-resistant and heat-sensitive variants, cells that are resistant to oxidative stress and in cells exposed to chemical treatments that protect or sensitize to heat-induced cells killing (summarized in ).
Table I. Summary of the relationship of heat sensitivity and the heat induced γ-H2AX response.
The attenuation of the heat-induced γ-H2AX response in transiently TT cells was found when clonogenic thermotolerance was induced by heat but not by sodium arsenite. Heat-induced thermotolerance has a component that is independent of protein synthesis while the arsenite induced one is not Citation11. Since the attenuation of the γ-H2AX response is observed in cells that recover from a thermotolerance-inducing heat treatment in the presence of cycloheximide, the attenuation of the γ-H2AX response in heat-induced transiently thermotolerant cells is probably associated with the PSI state of thermotolerance and not associated with the increased levels of heat shock proteins. In order to further test the notion that the heat-induced γ-H2AX response plays a role in heat-induced cell killing, we took advantage of a collection of mutants and variants of Chinese hamster cells that have altered heat sensitivities due to different mechanisms. We asked two questions. First, is the initial magnitude of the γ-H2AX response found in cells of different heat sensitivities the same as found in their wild type counterparts, and second, is the resolution of the heat-induced γ-H2AX foci modified in parallel with heat sensitivity. We have shown earlier that with respect to heat-induced alterations in several cellular parameters involved in cell killing by heat, the initial magnitude of the perturbations did not correlate with thermal sensitivity, while the recovery from such perturbations did Citation21, Citation36. We found that the initial γ-H2AX response was not attenuated in any of the cell lines we examined and that the resolution of the heat-induced γ-H2AX foci was independent of thermal sensitivity. Furthermore, the γ-H2AX response was not modified in cells treated with agents that protect or sensitize cells to hyperthermic cell killing such as glycerol and amino acid analogs. We also found that while H2AX knock-out cells are radiosensitive, they are not heat sensitive. Thus, we have demonstrated using several different approaches, that the heat-induced γ-H2AX response is not associated with a process that plays a role in thermal sensitivity and by implication, in heat-induced cell killing.
Exposure of cells to elevated temperatures induces oxidative stress, generating free radicals that have been reported to induce DNA base damage Citation37. A mechanism for the generation of additional DNA DSB in heated-irradiated cells through interference with the repair of base damage by the nucleotide excision repair pathway has been proposed Citation38, Citation39. It has been hypothesized that heat-induced oxidative stress is involved in such a mechanism in cells exposed to heat alone Citation29. Therefore, we also ascertained the heat-induced γ-H2AX response in an oxidative stress-resistant cell line that overexpresses catalase and glutathione Citation31, Citation32. However, neither the heat-induced γ-H2AX response nor its resolution was altered in this cell line when compared to its wild-type counterpart, a result that is not consistent with the notion that heat-induced oxidative stress leads to the generation of DNA DSB Citation29.
The original report on the heat-induced γ-H2AX response proposed that heat induces γ-H2AX foci through heat-induced protein damage Citation4, a process that is known to be involved in hyperthermic cell killing Citation15, Citation40, Citation41. Several of our results argue against this possibility. First, molecular chaperones, that are known to attenuate heat-induced proteotoxicity, do not protect against induction of γ-H2AX foci by heat, since over-expression of either hsc70 or hsp27 did not attenuate either the initial magnitude of, or recovery from, the heat-induced γ-H2AX response. Second, the thermotolerance for the γ-H2AX response is only observed in heat-induced, but not sodium arsenite-induced thermotolerance. The heat shock proteins induced by sodium arsenite are the same as are induced by heat shock Citation16, Citation19, Citation42. Third, the observation that the heat-induced γ-H2AX response is attenuated in cells that recover from a thermotolerance-inducing heat treatment in the presence of cycloheximide, the so-called PSI state of thermotolerance, indicates that elevated expression of molecular chaperones is not required for an attenuation of the γ-H2AX response. Fourth, neither the induction nor the resolution of heat-induced γ-H2AX foci was affected in hsp70 knockout cells Citation6. Last, the heat protector glycerol has been shown to protect against proteotoxicity Citation24–26, yet the γ-H2AX response is not attenuated in glycerol treated cells. Overall, our results argue against a role for heat-induced proteotoxicity in the γ-H2AX response observed in heated cells.
The mechanism by which heat induces γ-H2AX foci still remains unclear. We have shown that such induction is dependent on ataxia-telangiectasia mutated protein (ATM) and that heat activates ATM Citation6. Thus, the involvement of heat-induced chromatin structure alterations in this process is an attractive hypothesis. The only set of experiments that have examined whether global chromatin structure alterations can lead to the formation of γ-H2AX foci have used hypotonic treatments for 1 h, chloroquine for 4 h or trichostatin for 8 h and found that such treatments did not induce γ-H2AX Citation43. On the other hand, γ-H2AX foci are observed immediately after heating with as little as 15 min at 43°C. Therefore, heat may induce a rapid transitory chromatin alteration which allows the heat-activated ATM to phosphorylate chromatin-associated H2AX molecules. The resolution of the γ-H2AX foci would then involve the dephosphorylation of γ-H2AX and/or its removal by exchange, as has been shown for ionizing radiation Citation44, Citation45, since the time course of resolution of the majority of γ-H2AX is similar in heated and irradiated cells [see this paper and Citation46. Recently it has been reported that the DNA damage response, including the induction of γ-H2AX foci, can be induced in the absence of DNA lesions Citation47. This was demonstrated by tethering MDC1 to chromatin to sites of a transfected bacterial gene, an interaction that resulted in the formation of γ-H2AX foci. We have shown that heat also induces MDC1 foci Citation6. Thus, heat may lead to a tethering or interaction of MDC1with chromatin that leads to the formation of γ-H2AX foci. This possibility appears to be likely because heat has been shown to induce new interactions of nuclear proteins with chromatin Citation48. Thus, heating may result in the interaction of MDC1 with chromatin, either transitory or permanent, leading to the formation of γ-H2AX. MDC1 has been shown to exchange very rapidly in its interaction with chromatin in non-treated cells Citation49. Thus, as an alternate or complementary mechanism for the formation of γ-H2AX foci, heat may lead to an increase in the residence time of MDC1 on chromatin, resulting in the formation of γ-H2AX foci. The putative heat-induced alterations in chromatin that result in the formation of γ-H2AX foci may represent a new and novel effect of heat on cellular structures such as chromatin, the mechanism of which remains to be elucidated.
Acknowledgements
This research was supported by P01-CA104457 and funds the Department of Radiation Oncology. We thank Dr. J Roti Roti for valuable comments on the manuscript.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: The histone guardian of the genome. DNA Repair (Amst) 2004; 3: 959–967
- Takahashi A, Ohnishi T. Does gammaH2AX foci formation depend on the presence of DNA double strand breaks?. Cancer Lett 2005; 229: 171–179
- Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res 2002; 158: 486–492
- Takahashi A, Matsumoto H, Nagayama K, Kitano M, Hirose S, Tanaka H, Mori E, Yamakawa N, Yasumoto J, Yuki K, et al. Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 2004; 64: 8839–8845
- Kaneko H, Igarashi K, Kataoka K, Miura M. Heat shock induces phosphorylation of histone H2AX in mammalian cells. Biochem Biophys Res Commun 2005; 328: 1101–1106
- Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, Gupta A, Rief N, Horikoshi N, Baskaran R, et al. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res 2007; 67: 3010–3017
- Ohtsuka K, Laszlo A. The relationship between hsp70 localization and heat resistance. Exp Cell Res 1992; 202: 507–518
- Riballo E, Kuhne M, Rief N, Doherty A, Smith G, Reico M, Reis C, Dahm K, Fricke A, Krempler A, et al. A pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to gamma-H2AX foci. Mol Cell 2004; 16: 715–724
- Ward I, Kim JE, Minn K, Chini CC, Mer G, Chen J. The tandem BRCT domain of 53BP1 is not required for its repair function. J Biol Chem 2006; 281: 38472–38477
- Laszlo A, Davidson T, Harvey A, Sim J, Malyapa R, Spitz D, Roti Roti J, et al. Alterations in heat-induced radiosensitization accompanied by nuclear structure alterations in Chinese hamster cells. Int J Hyperthermia 2006; 22: 43–60
- Laszlo A. Evidence for two states of thermotolerance in mammalian cells. Int J Hyperthermia 1988; 4: 513–526
- Borrelli MJ, Stafford DM, Karczewski LA, Rausch CM, Lee YJ, Corry PM. Thermotolerance expression in mitotic CHO cells without increased translation of heat shock proteins. J Cell Physiol 1996; 169: 420–428
- Laszlo A. The thermoresistant state: Protection from initial damage or better repair?. Exp Cell Res 1992; 202: 519–531
- Stege GJ, Brunsting JF, Kampinga HH, Konings AW. Thermotolerance and nuclear protein aggregation: Protection against initial damage or better recovery?. J Cell Physiol 1995; 164: 579–586
- Kampinga HH. Thermotolerance in mammalian cells. Protein denaturation and aggregation, and stress proteins. J Cell Sci 1993; 104: 11–17
- Kampinga HH, Brunsting JF, Stege GJ, Burgman PW, Konings AW. Thermal protein denaturation and protein aggregation in cells made thermotolerant by various chemicals: Role of heat shock proteins. Exp Cell Res 1995; 219: 536–546
- Chen MS, Featherstone T, Laszlo A. Amplification and altered expression of the hsc70/U14 snoRNA gene in a heat resistant Chinese hamster cell line. Cell Stress Chaperones 1996; 1: 47–61
- Landry J, Chretien P, Lambert H, Hickey E, Weber LA. Heat shock resistance conferred by expression of the human HSP27 gene in rodent cells. J Cell Biol 1989; 109: 7–15
- Laszlo A, Li GC. Heat-resistant variants of Chinese hamster fibroblasts altered in expression of heat shock protein. Proc Natl Acad Sci USA 1985; 82: 8029–8033
- Laszlo A. The relationship of heat-shock proteins, thermotolerance, and protein synthesis. Exp Cell Res 1988; 178: 401–414
- Laszlo A, Davidson T, Hu A, Landry J, Bedford J. Putative determinants of the cellular response to hyperthermia. Int J Radiat Biol 1993; 63: 569–581
- Harvey WF, Bedford JS. Isolation and characterization of heat-sensitive, thermotolerant defective mutants of CHO cells. Radiat Res 1988; 113: 526–542
- Henle KJ, Warters RL. Heat protection by glycerol in vitro. Cancer Res 1982; 42: 2171–2176
- Pittet JF, Lee H, Pespeni M, O'Mahony A, Roux J, Welch WJ. Stress-induced inhibition of the NF-kappaB signaling pathway results from the insolubilization of the IkappaB kinase complex following its dissociation from heat shock protein 90. J Immunol 2005; 174: 384–394
- Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones 1996; 1: 109–115
- Kampinga HH, Turkel-Uygur N, Roti Roti JL, Konings AW. The relationship of increased nuclear protein content induced by hyperthermia to killing of HeLa S3 cells. Radiat Res 1989; 117: 511–522
- Li GC, Laszlo A. Amino acid analogs while inducing heat shock proteins sensitize CHO cells to thermal damage. J Cell Physiol 1985; 122: 91–97
- Laszlo A, Li GC. Effect of amino acid analogs on the development of thermotolerance and on thermotolerant cells. J Cell Physiol 1993; 154: 419–432
- Takahashi A, Ohnishi T. What is the critical hyperthermia target in cancer cells. Jpn J Hyperthermic Oncol 2006; 22: 229–237
- Spitz DR, Li GC, McCormick ML, Sun Y, Oberley LW. Stable H2O2-resistant variants of Chinese hamster fibroblasts demonstrate increases in catalase activity. Radiat Res 1988; 114: 114–124
- Spitz DR, Adams DT, Sherman CM, Roberts RJ. Mechanisms of cellular resistance to hydrogen peroxide, hyperoxia, and 4-hydroxy-2-nonenal toxicity: The significance of increased catalase activity in H2O2-resistant fibroblasts. Arch Biochem Biophys 1992; 292: 221–227
- Spitz DR, Kinter MT, Roberts RJ. Contribution of increased glutathione content to mechanisms of oxidative stress resistance in hydrogen peroxide resistant hamster fibroblasts. J Cell Physiol 1995; 165: 600–609
- Spitz DR, Li GC. Heat-induced cytotoxicity in H2O2-resistant Chinese hamster fibroblasts. J Cell Physiol 1990; 142: 255–260
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow S, Fleming J, Monroe B, Ciccone D, Yan C, Vlaskova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Nat Acad Sci USA 2002; 99: 8173–8178
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen H, Sedelnikova O, Reina-San-Martin B, Coppola V, Meffre E, et al. Genomic instability in mice lacking histone H2AX. Science (New York) 2002; 296: 922–927
- Laszlo A. The effects of hyperthermia on mammalian cell structure and function. Cell Prolif 1992; 25: 59–87
- Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res 2002; 30: 1354–1363
- Kampinga HH, Dikomey E. Hyperthermic radiosensitization: Mode of action and clinical relevance. Int J Radiat Biol 2001; 77: 399–408
- Kampinga HH, Dynlacht JR, Dikomey E. Mechanism of radiosensitization by hyperthermia (≥43°C) as derived from studies with DNA repair defective mutant cell lines. Int J Hyperthermia 2004; 20: 131–139
- Lepock JR. How do cells respond to their thermal environment?. Int J Hyperthermia 2005; 21: 681–687
- Roti Roti JL, Kampinga HH, Malyapa RS, Wright WD, vanderWaal RP, Xu M. Nuclear matrix as a target for hyperthermic killing of cancer cells. Cell Stress Chaperones 1998; 3: 245–255
- Li G. Induction of thermotolerance and enhanced heat shock protein synthesis in Chinese hamster fibroblasts by sodium arsenite and ethanol. J Cell Phys 1983; 115: 116–122
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421: 499–506
- Kusch T, Workman JL. Histone variants and complexes involved in their exchange. Sub Cell Biochem 2007; 41: 91–109
- Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell 2005; 20: 801–809
- Mirzayans R, Severin D, Murray D. Relationship between DNA double-strand break rejoining and cell survival after exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: Implications for evaluation of clinical radiosensitivity. Int J Radiat Oncol Biol Phys 2006; 66: 1498–5105
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science Express 2008; 320: 1507–1510
- Roti Roti JL. Cellular responses to hyperthermia (40–46°): Cell killing and molecular events. Int J Hyperthermia 2008; 24: 3–15
- Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson S, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 2004; 23: 2674–2683