Abstract
Purpose: A proof-of-concept study to evaluate a new autofluorescence method to differentiate necrotic thermally fixed cells from viable tissue following thermal ablation.
Methods: A conductive interstitial thermal therapy (CITT) device was used to ablate swine mammary tissue and rabbit VX-2 carcinomas in vivo. The ablated regions and 10-mm margins were resected 24 h following treatment, embedded in HistOmer® and sectioned at 3 mm. The fresh sections were evaluated for gross viability with triphenyl tetrazolium chloride, 1 h post-resection. Representative non-viable and viable areas were then processed and embedded into paraffin, and sectioned at 5 µm. Standard H&E staining and proliferating cell nuclear antigen (PCNA) immunohistochemistry were compared against autofluorescence intensity, at 488-nm wavelength, for cellular viability.
Results: Heat-fixed cells in non-viable regions exhibit increased autofluorescence intensity compared to viable tissue (area under receiver operating characteristics (ROC) curve = 0.96; Mann-Whitney P < 0.0001). An autofluorescence intensity-based classification rule achieved 92% sensitivity with 100% specificity for distinguishing non-viable from viable samples. In contrast, PCNA staining did not reliably distinguish heat-fixed, dead cells from viable cells.
Conclusions: Examination of H&E-stained sections using autofluorescence intensity-based classification is a reliable and readily available method to accurately identify heat-fixed cells in ablated surgical margins.
Introduction
The recent emergence of heat-based therapies has engendered many advocates for its use to locally control and treat solid cancerous tumours Citation[1–3]. Many techniques have been developed to heat the diseased tissue above a threshold temperature to induce irreversible thermal damage. Radiofrequency ablation (RFA), laser, interstitial and external ultrasound devices and, recently, conductive interstitial thermal therapy (CITT) are just a few of the available technologies where thermal ablation has been shown to be effective in destroying solid tumours and margins Citation[4–11]. To date, the efficacy of thermal ablation techniques has been determined by employing ablate-resect techniques Citation[10],Citation[12–16]. In ablate-resect procedures, histopathological examination is the standard method to determine the outcomes of the thermal ablation treatment. Pathologically there are three zones of tissue damage within the ablated zone: a central heat-fixed region surrounded by a zone of coagulative necrosis, and an outer rim of mixed viable tissue and necrotic tissue Citation[17–20]. Cells in the heat-fixed region retain many of the morphological and immunohistochemical characteristics of viable cells Citation[17],Citation[20], and thus are difficult to identify as non-viable in histological sections.
There is no standard and well-accepted technique for evaluating the viability of cells in thermally ablated regions. There are several pathologic techniques currently used to evaluate non-viable cells in tissue sections. Supravital stains such as that to identify nicotinamide adenine dinucleotide (NADH) diaphorase can only be performed on fresh frozen sections. Immunohistochemistry against various cellular proteins may lead to misclassification of tissues because some antigens routinely used in immunohistochemical staining, such as proliferating cell nuclear antigen (PCNA), may be preserved in heat-fixed tissues in a manner similar to that seen in microwave fixation Citation[20]. Transferase-mediated dUTP nick end labelling (TUNEL) staining to detect apoptosis has a relatively low sensitivity which is reported to vary between 60% to 90%. Citation[21] The triphenyl tetrazolium chloride (TTC) viability stain can only be used in gross specimens because the red formazan fades during histological processing. Therefore, a simple and reliable method to accurately identify heat-fixed (dead) cells and avoid false-positive diagnoses of viability in histological specimens would be of great use in the growing field of thermal ablation of tumours.
Necrotic cells are known to exhibit increased autofluorescence when excited with light at 488 nm Citation[22–25], as cellular autofluorescence increases with decreasing metabolic activity Citation[26]. The exact mechanism of autofluorescence has not been elucidated, but it is thought to be due to increased levels of denatured proteins Citation[27],Citation[28]. An additional mechanism may be intracellular release of metabolites such as flavin adenine dinucleotide (FAD) or NADH from mitochondria Citation[29]. FAD is a likely candidate for this phenomenon, because it fluoresces at this wavelength Citation[26]. Some investigators have used this difference in autofluorescence intensity to identify and quantify necrosis in tumour sections, including those that are formalin-fixed, paraffin-embedded and routinely stained Citation[30],Citation[31]. To the best of our knowledge, it has not yet been determined that heat-fixed cells will exhibit significantly higher autofluorescence intensity in comparison to viable cells. Our objective was to determine the accuracy and reliability of a modified autofluorescence procedure, which we present herein, in differentiating heat-fixed cells from viable cells in thermally ablated regions. In addition, we compared this procedure to PCNA staining in identification of heat-fixed cells.
In this study we used CITT, an invasive thermal conductance ablation approach, to ablate VX2 rabbit carcinomas and pig mammary tissue to investigate tissue necrosis. It is important to bear in mind that a CITT device delivers the heat to the target tissue by conduction, thus the tissue is not exposed to electromagnetic field that could have potential confounding effects on cell viability. Thus, in CITT the thermal damage is only a function of the temperature attained and time exposed at any given point in the tissue (thermal dose).
Methods
Thermal ablation
The CITT device was used to ablate soft tissue and tumours in animal models in vivo. The principles of CITT have been described in detail elsewhere Citation[32],Citation[33]. Briefly, the CITT device consists of a thermal probe with deployable pins and computer controlled heater that allows high-dose heat delivery to the target tissue. Thermal ablation was conducted while the animals were under general anaesthesia. The animal experiments were approved by the UAMS Institutional Animal Care and Use Committee and were conducted according to the guidelines in the Guide for the Care and Use of Laboratory Animals, NIH Publication No. 86-23. Tissues examined were from two separate ablation studies using similar techniques, but with slight differences in thermal dose.
Rabbit carcinoma: Fresh VX2 carcinoma (National Cancer Institute, Frederick Cancer Center, Frederick, MD) was implanted intramuscularly in the rear leg of 8 adult female NZW rabbits. The tumours were allowed to grow to 2–3 cm diameter, whereupon they were ablated for 20 min at the maximum probe temperature of 120°C, in vivo. Citation[32]
Pig mammary gland: The female pig model was selected because it is an accepted model for studying interstitial thermal therapy for thermal ablation in human-like breast tissue Citation[34],Citation[35]. In this study, ablations were performed, in vivo, on tissue under 8 nipples in an adult, non-pregnant female at various temperatures within the range of 79°C to 170°C and total ablation times of 17 to 25 min Citation[33].
Tissue preparation: Following ablation, the treated region and approximately 5–10 mm of adjacent tissue were resected and placed in buffered saline at 2°C for 30 min. Thereafter, the entire excised tissue was embedded in HistOmer (Vibratome, St Louis, MO) for 5 min and sliced into 3-mm sections perpendicular to the ablated surface. The slices were stained for viability with TTC 1% solution for 1 h at 37°C. In viable tissue, TTC is reduced to red formazan by the mitochondrial enzyme succinate dehydrogenase. This reaction therefore only occurs in viable tissue. Following TTC staining, the tissue was removed from the solution, and sections were photographed with a digital camera (Canon ES Rebel 350). The tissue specimens were removed from HistOmer and fixed in 10% neutral buffered formalin. After fixation, samples measuring approximately 1 cm × 1 cm were chosen from TTC-positive (red-stained) and TTC-negative (unstained) areas, and these samples were processed and embedded into paraffin, sectioned at 5 µm, and routinely stained with haematoxylin and eosin (H&E). Additionally, 5 samples of normal rabbit skin from non-ablated sites and 5 samples of non-ablated pig skin were processed without TTC staining.
Histological evaluation
Digital threshold
We devised a simple method to aid in diagnosis of viable vs. non-viable areas and cells in multiple sections, using a digital filter to eliminate background autofluorescence. Digital images of fluorescent sections were converted to greyscale in Image J, and a threshold intensity value was set using the ‘image-adjust-threshold’ menu to exclude background fluorescence of normal epithelial structures in a non-ablated control sample of pig mammary gland and skin. Using the black and white method of digital mark-up, normally fluorescent structures (collagen and keratin) remain white, while viable epithelial cells and tumour cells are black. The threshold settings were applied to normal epithelial and stromal cells in 5 control pig skin samples. This method was repeated on rabbit skin and mammary tissue using 5 normal rabbit samples from 5 different rabbits. For direct comparison of fluorescence and cellular morphology, fluorescent photos were overlaid onto H&E photos using Adobe Photoshop CS2 layers function. We next applied this threshold value to images of thermally ablated sections of VX-2 carcinomas and pig mammary tissue.
Quantitative analysis. To verify that necrotic and heat-fixed cells exhibit increased autofluorescence intensity compared to viable cells, samples of rabbit VX-2 carcinoma and pig mammary tissue and skin were compared. Samples from VX2 carcinoma were taken from TTC positive (viable, n = 5) regions and from TTC negative (non-viable, n = 25) regions. Additionally, 3 samples of control (non-CITT-treated) skin and mammary tissue from 3 different pigs were examined. H&E slides were examined under bright field microscopy and using fluorescence microscopy at 488 nm wavelength (FITC channel) using a Nikon Eclipse 2000E microscope attached to a Nikon digital camera. Camera and lamp settings were held constant for all sections. Serial photos of each section were taken from the same microscopic 200 x field using both bright field and fluorescence microscopy. Unmodified photos were analysed using NIH Image J software. Within each image, five cells, located at approximately 12:00, 2:00, 5:00, 8:00, and 10:00 on the clock face were outlined using the Polygon function and individually analysed. Each cell was delineated, and its autofluorescence intensity was determined as the distributional mean of the cell's fluorescence greyscale intensities using the Histogram function; the resulting intensities of the five cells were then averaged and taken as representative of the intensity of cells within the tissue-section image. Fluorescence intensities of individual tissue sections were plotted along with group means and standard deviations. Intensity distributions were assessed for degree of separation between the two groups via receiver operating characteristics (ROC) analysis; the area under the ROC curve (the ROC AUC) was compared to the null-hypothesis value 0.50 (denoting complete overlap of distributions) via the one-sided Mann-Whitney test. The point on the empirical ROC curve that minimised misclassification error (by maximising the sum of sensitivity and specificity) was used to identify the upper and lower limits on a range of intensity thresholds that would have optimal classification characteristics. Statistical analyses were performed using Excel 2003 (Microsoft Corporation, Redmond, WA) and NCSS 2004 (Number Cruncher Statistical Systems, Kaysville, UT).
Immunohistochemistry. To determine whether or not heat-fixed cells may retain reactivity to anti-PCNA antibodies, we stained sections from TTC-positive and TTC-negative regions of VX-2 carcinoma, normal mammary gland, and skin from rabbit and pig. Sections were deparaffinised and rehydrated through a graded series of alcohols and water. Antigen retrieval was performed in a Dako decloaker (Biocare Medical, Walnut Creek, CA) for 20 min using Dako target retrieval (pH 6). After rinsing for 3 min in distilled water, Dako hydrogen peroxidase was applied to the sections for 10 min, and slides were rinsed in Dako tris buffered saline tween-20 (TBST). Sections were incubated for 30 min in 10% normal goat serum protein block diluted in TBST at room temperature (RT), and then blotted. Sections were then incubated in primary antibody (PCNA, PC-10, mouse monoclonal, predilute, Abcam ab912) for 1 h at RT and rinsed in TBST. Sections were incubated in secondary antibody (biotinylated goat anti-mouse, Vector Labs, Burlingame, CA) diluted 1:400 in TBST for 30 min at RT and rinsed in TBST. Sections were incubated in Vector Elite ABC (Vector Labs) for 30 min at RT and rinsed in TBST. Dako DAB + 3 was applied for 3 min; sections were rinsed in TBST, lightly counterstained with haematoxylin, mounted, examined under a light microscope, and representative regions were digitally photographed at 200× magnification as described above.
Results
Digital threshold: Based on autofluorescence intensity, we were consistently able to digitally exclude viable cells in control sections of pig and rabbit skin and mammary gland with a single Image J threshold setting. While cellular layers of the epidermis exhibit minimal autofluorescence intensity, strong autofluorescence is present in keratin and dermal collagen (). The digital threshold was set to exclude autofluorescence from viable epidermal cells, which remain black in the filtered image (). This threshold setting also excluded viable cells in mammary epithelium and hair follicles (). Non-viable cells in ablated skin and VX-2 carcinomas remain white in filtered images ().
Figure 1. (A) Normal pig skin. Viable epidermal cells (star) exhibit minimal autofluorescence that is excluded (black) by the ImageJ filter (C). Dermal collagen (arrow) and superficial keratin are not excluded by the filter and remain white. H&E (A), fluorescent (B), and filtered (C) 400×. (B) Normal pig skin. Viable hair follicle (A,C,E) and mammary gland (B,D,F). Follicular and mammary epithelia (arrows) exhibit minimal autofluorescence that is excluded by the filter. H&E (A,B), fluorescent (C,D) and filtered (E, F) 400× magnification.
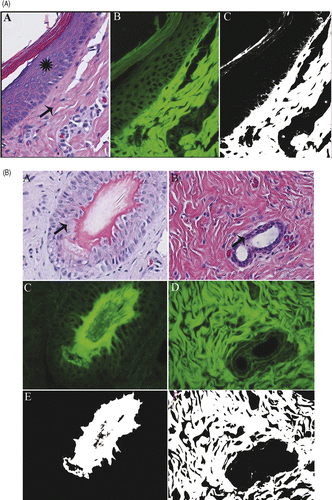
Figure 2. Tumor and skin ablation: Non-viable, ablated skin (A,B,C) and rabbit VX2 carcinoma (D,E,F). Ablated non-viable skin (A,B,C) exhibits increased autofluorescence, and remains white in filtered image. Non-viable tumor cells are strongly autofluorescent, and are white in filtered image. Viable VX2 carcinoma cells (G,H,I) fluoresce less strongly and remain black in filtered image. H&E (A,D,G), fluorescent (B,E,H), filtered (C,F,I), 400× magnification.
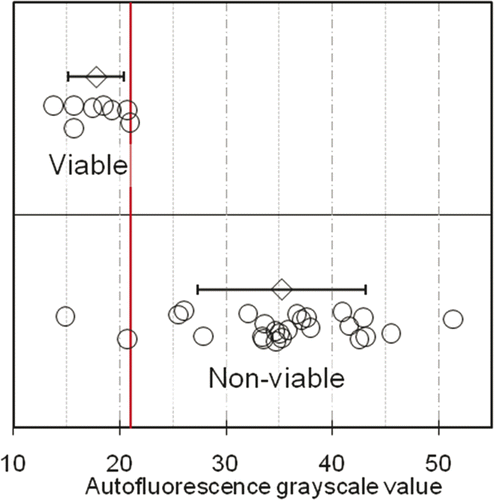
Quantitative analysis: Cells in regions verified as heat-fixed or necrotic via TTC staining exhibited marked increase in autofluorescence intensity over viable epithelial and tumour cells. The average intensity of autofluorescence in cells within these regions was significantly increased, with a mean ± SD greyscale value of 35.2 ± 7.9 for non-viable cells compared to 17.8 ± 2.6 for viable cells. ROC analysis was used to assess the two groups’ autofluorescence-intensity distributions for discriminative potential, with results shown in . The area under the empirical ROC curve was 0.96 (Mann-Whitney U = 192, 1-sided P = 5.6 × 10−5), indicating very little overlap between the distributions of viable and non-viable samples, and thus very good potential to classify them correctly using an autofluorescence intensity (greyscale) threshold. The ROC curve of shows how the sensitivity and specificity of such a classification rule changes with different settings of the greyscale threshold. The classification rule is most accurate when the sum of sensitivity and specificity reaches a maximum, which occurs for this data at 92% sensitivity and 100% specificity (red ellipse in ). This optimal combination of sensitivity and specificity is attained when the greyscale threshold is located somewhere between 20.99 and 25.49. shows how the samples are classified when the greyscale threshold (vertical red line) is chosen for convenience to have an integer value of 21. All eight of the viable samples have their fluorescence intensities below the threshold (100% specificity), while 23 of the 25 samples have intensities above the threshold (92% sensitivity). Moving the threshold up to 25.49, or down to 20.99, does not change which samples fall below the threshold and which remain above it.
Figure 3. Receiver Operating Characteristics (ROC) analysis of using an autofluorescence intensity (grayscale) threshold to distinguish viable from non-viable samples. The line at top is the empirical ROC curve for classifying Fig 4 data via grayscale threshold; the diagonal is the theoretical ROC curve of a classification rule with no discriminatory potential. The area under the empirical ROC curve is 0.96 (1-sided Mann-Whitney P = 5.6 × 10−5). The ellipse denotes where sensitivity (92%) and specificity (100%) has a sum that is maximized. This optimal combination of 92% sensitivity with 100% specificity is produced by a range of grayscale intensity thresholds that have a minimum of 20.99 and maximum of 25.49.
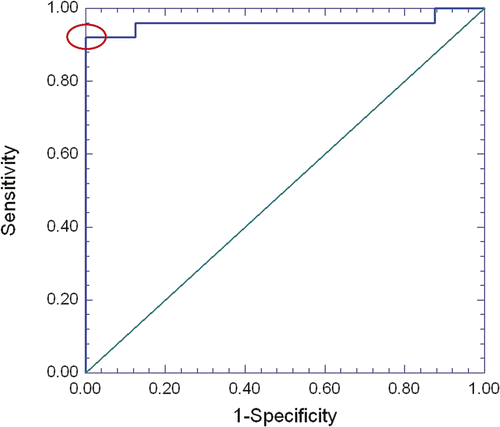
Figure 4. Distribution of autofluorescence grayscale values of viable (n = 5 VX2 v samples and n = 3 non-ablated pig skin samples) and non-viable (heat-killed, n = 25 VX2 carcinoma samples) tissue sections. Open circles denote tissue-section fluorescence intensities, determined as the average grayscale value of single cells in five pre-determined locations in the tissue section; grey diamonds (error bars) denote the sample averages (standard deviations) of fluorescence intensities of the viable and non-viable sections. The vertical line denotes an integer-valued grayscale threshold chosen from the optimal range of thresholds identified by ROC analysis (Fig 3 legend). 23 out of 25 non-viable samples lie above the threshold (92% sensitivity), while all 8 viable samples lie below the threshold (100% specificity).
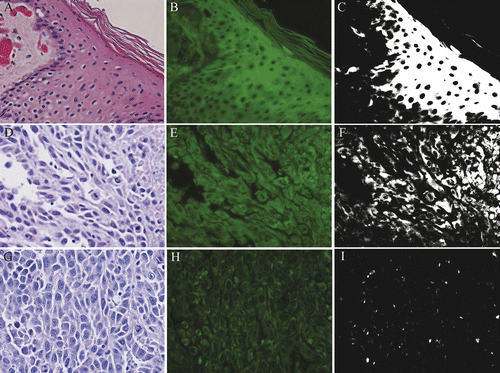
By contrast, PCNA was not reliable as an indicator of cell viability. Labelling of nuclei in TTC-negative, non-viable regions was less intense than in most TTC-positive, viable regions, but was often present in areas that were non-viable as assessed by TTC and by autofluorescence imaging ().
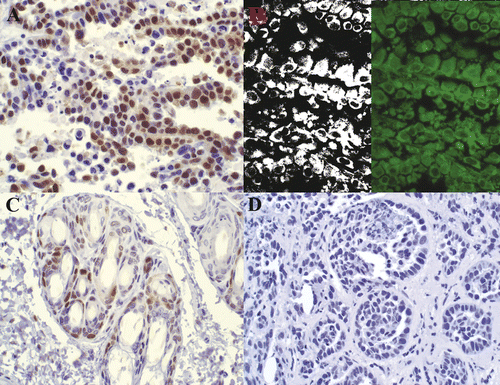
Figure 5. PCNA staining in non-viable tumor cells. Rabbit VX2 carcinoma. Strong nuclear PCNA antibody labeling (A) is observed in tumor cells within a region identified as non-viable via TTC. Fluorescent and filtered images (B) demonstrate strong autofluorescence. Control (C) with primary antibody omitted is unstained. 400× magnification.
Discussion
Complete coagulation and necrosis of the target tumour and margins is a prerequisite for a successful thermal ablation procedure. It is equally important to avoid unnecessary damage to healthy tissue to minimise morbidity. Thus, it is imperative to precisely determine the exact size of the thermally ablated (dead) region as well as the optimal time-course with which to assess the extent of ablation so that more consistent and more effective ablation strategies can be developed.
Fixation of cells in situ causes a problem for the pathologist who must determine that surgical margins are free of viable tumour. Cell death via necrosis or apoptosis is accompanied by consistent morphological changes. Necrotic cells are generally swollen, may stain more intensely with eosin, and contain ruptured and fragmented nuclei. Apoptotic cells are shrunken with small, dense nuclei or nuclear remnants. In contrast, the process of fixation inactivates the cellular enzymes responsible for morphologic features of necrosis or apoptosis. Therefore, heat-fixed cells retain the morphology of viable cells for long periods of time, without features of apoptosis or necrosis. For instance, tissues prepared by microwave fixation, which relies on the fixative properties of heat, are comparable to formalin-fixed tissues for microscopic evaluation.
Reliable discrimination of viable from non-viable cells in margins has previously been limited to the use of frozen samples for labelling with enzymatic vital stains such as reduced NADH. In this work we tested the efficacy of autofluorescence intensity-based method to delineate heat-fixed thermally ablated cells from viable non-ablated cells in formalin-fixed and paraffin-embedded samples. We based our approach on the fact that differences in autofluorescence intensity have been reliably used to rapidly diagnose and quantify necrosis in tumour sections treated with non-thermal agents Citation[30],Citation[31]. Our results here demonstrate that autofluorescence is also diagnostically useful in examining thermally ablated tissue. In addition, our findings, albeit in a small number of samples, suggest that autofluorescence intensity may be more sensitive than PCNA staining to discriminate between viable and non-viable cells in these ablated tumours. Staining for PCNA was preserved in heat-fixed tissues, indicating a false positive for cell survival. Although we did observe differences in the intensity of PCNA staining between heat-fixed and viable cells, there was not complete loss of staining in heat-fixed areas. One possible explanation for this finding is that PCNA reactivity is retained in heat-fixed cells because heat-fixation does not lead to protein cross-linking. All tissues in this study were placed into formalin, but heat-fixation may have provided stabilisation of protein and resistance to formalin-induced protein cross-linking.
In contrast, using a digital threshold to eliminate background levels of mammary and epidermal autofluorescence, we were able to reliably detect heat-fixed (dead) cells in regions verified as non-viable by TTC staining. Moreover, using an autofluorescence greyscale threshold between 20.99 and 25.49, we correctly classified all our viable samples (100% specificity) and achieved 92% sensitivity, area under ROC curve = 0.96; Mann-Whitney P < 0.0001. It should be noted that the range of optimal greyscale thresholds we obtained from ROC analysis were specific to our samples and the properties of our microscope, and that different laboratories will undoubtedly want to determine their own range of optimal thresholds.
Interstitial collagen and epidermal keratin do not affect this analysis because only tumour and epithelial cells were selected for histogram analysis. In only two samples was there a noticeable discrepancy between the TTC staining and autofluorescence measurements, where viable cells were not identified in the TTC staining. Other researchers have demonstrated that TTC staining does not affect fluorescence properties of cells Citation[36], therefore the staining procedure should not have affected autofluorescence.
It is possible that cells were misclassified by the autofluorescence method; however, in these samples the levels of autofluorescence were extremely low, and were consistent with the levels observed from cells in TTC-viable sections. It is also unlikely that macroscopic areas of tissue would be misclassified by TTC staining because it is dependent on functioning mitochondrial enzymes to produce red formazan. Therefore, we surmise that the existence of small foci of viable cells went undetected in these cases in the TTC section because the TTC staining is examined without magnification.
The described method identifies viable cells in ablated margins. Strongly autofluorescent structures such as collagen and keratin are present in normal skin and connective tissue surrounding tumours. The image analysis digital threshold is set to exclude background autofluorescence of viable cells and strongly autofluorescent stromal material. In this way, viable cells are identified (remain black) in contrast to more strongly fluorescent collagen, keratin, and dead tumour cells; all of which are more uniformly autofluorescent than viable tumour. Using such a filter, viable tumour cells in ablated margins may be identified by a pathologist in a single routinely stained slide without the confounding evaluation of serial sections. In margin evaluation, the presence of viable tumour cells indicates that further treatment may be necessary. Although we did not evaluate the effects of ablation on autofluorescence of collagen, future studies may include defining the fluorescence of ablated stromal tissue as well, since mounting evidence indicates that recurrent or metastatic cancer relies on the stromal environment. This method could be expanded to reliably identify and quantify regions of necrosis and heat fixation with the addition of a second filter to eliminate the strong autofluorescence of collagen and keratin.
A major benefit of the autofluorescence method we presented herein is that it may be applied to individual biopsy specimens. We have demonstrated that it sufficed to set a digital threshold from a single non-ablated control sample of mammary epithelium and skin to identify viable and non-viable cells in multiple sections of a mammary carcinoma (VX2) and skin from different animals. Thus, digital threshold parameters (or threshold values) can be set once and applied to multiple sections. Although we have only validated our autofluorescence method in related epithelial tissues (VX2 carcinoma and rabbit and porcine mammary tissue) that are commonly used to study thermal ablation, we expect that our method may be useful in other types of thermally ablated animal or human tissues as well. Because different types of cells exhibit varying degrees of normal autofluorescence intensity due to metabolic and structural differences, the method must be validated individually in other tissue types. Additionally, because different laboratories have different microscopes, the classification threshold will undoubtedly need to be calibrated to slightly different set-points for different labs to achieve optimal performance in each. But once established and calibrated, a single digital threshold may suffice to identify non-viable regions from a range of samples from similar organs or tumours.
Conclusions
In this work we demonstrated the efficacy of autofluorescence intensity-based classification to delineate heat-fixed thermally ablated cells from viable cells in formalin-fixed and paraffin-embedded samples using instruments and software that are readily accessible to a majority of pathologists. This simple and reliable method holds great promise as a means to reliably distinguish non-viable, heat-fixed cells from living tumour cells in the margins of ablated tumours. Validation in human cancer specimens and other research models appears warranted.
Acknowledgements
Funding for this work was provided by the National Cancer Institute NIH/NCI Grant No. CA108678-02 GS and CA44114 to RG. We would like to thank Dr Michael Borrelli for access to the fluorescent microscope and digital camera used in this work.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Crocetti L, Lencioni R. Thermal ablation of hepatocellular carcinoma. Cancer Imaging 2008; 8: 19–26
- Garrean S, Hering J, Saied A, Helton WS, Espat NJ. Radiofrequency ablation of primary and metastatic liver tumors: A critical review of the literature. Am J Surg 2008; 195: 508–520
- Pennathur A. Radiofrequency ablation for the treatment of lung neoplasm. Expert Rev Med Devices 2008; 5: 613–621
- Jeffrey SS, Birdwell RL, Ikeda DM, Daniel BL, Nowels KW, Dirbas FM, Griffey SM. Radiofrequency ablation of breast cancer: First report of an emerging technology. Arch Surg. 1999; 134: 1064–1068
- Izzo F, Thomas R, Delrio P, Rinaldo M, Vallone P, DeChiara A, Botti G, D'Aiuto G, Cortino P, Curley SA. Radiofrequency ablation in patients with primary breast carcinoma: A pilot study in 26 patients. Cancer 2001; 92: 2036–2044
- Akimov AB, Seregin VE, Rusanov KV, Tyurina EG, Glushko TA, Nevzorov VP, Nevzorova RF, Akimova EV. Nd: YAG interstitial laser thermotherapy in the treatment of breast cancer. Lasers Surg Med 1998; 22: 257–267
- Minhaj AM, Mann F, Milne PJ, Denham DB, Salas N, Jr, Nose I, Daamgard-Iversen K, Parel JM, Robinson DS. Laser interstitial thermotherapy (LITT) monitoring using high-resolution digital mammography: Theory and experimental studies. Phys Med Biol 2002; 47: 2987–2999
- Milne PJ, Parel JM, Manns F, Denham DB, Gonzalez-Cirre X, Robinson DS. Development of stereotactically guided laser interstitial thermotherapy of breast cancer: In situ measurement and analysis of the temperature field in ex vivo and in vivo adipose tissue. Lasers Surg Med 2000; 26: 67–75
- Vogl TJ, Mack MG, Roggan A, Straub R, Eichler KC, Muller PK, Knappe V, Felix R. Internally cooled power laser for MR-guided interstitial laser-induced thermotherapy of liver lesions: Initial clinical results. Radiology 1998; 209: 381–385
- Boehm T, Malich A, Reichenbach JR, Fleck M, Kaiser WA. Percutaneous radiofrequency (RF) thermal ablation of rabbit tumors embedded in fat: A model for RF ablation of breast tumors. Invest Radiol 2001; 36: 480–486
- Hazle JD, Stafford RJ, Price RE. Magnetic resonance imaging-guided focused ultrasound thermal therapy in experimental animal models: Correlation of ablation volumes with pathology in rabbit muscle and VX2 tumors. J Magn Reson Imaging 2002; 15: 185–194
- Boehm T, Malich A, Goldberg SN, Reichenbach JR, Hilger I, Hauff P, Reinhardt M, Fleck M, Kaiser WA. Radio-frequency tumor ablation: Internally cooled electrode versus saline-enhanced technique in an aggressive rabbit tumor model. Radiology 2002; 222: 805–813
- Boehm T, Malich A, Nahum Goldberg S, Reichenbach JR, Hilger I, Fleck M, Kaiser WA. Vacuum-assisted resection of malignant tumors with and without subsequent radiofrequency ablation: Feasibility of complete tumor treatment tested in an animal model. J Vasc Interv Radiol 2001; 12: 1086–1093
- Goldberg SN, Gazelle GS, Solbiati L, Rittman WJ, Mueller PR. Radiofrequency tissue ablation: Increased lesion diameter with a perfusion electrode. Acad Radiol 1996; 3: 636–644
- Bhowmick P, Coad JE, Bhowmick S, Pryor JL, Larson T, De La Rosette J, Bischof JC. In vitro assessment of the efficacy of thermal therapy in human benign prostatic hyperplasia. Int J Hyperthermia. 2004; 20: 421–439
- Bhowmick S, Swanlund DJ, Coad JE, Lulloff L, Hoey MF, Bischof JC. Evaluation of thermal therapy in a prostate cancer model using a wet electrode radiofrequency probe. J Endourol 2001; 15: 629–640
- Coad JE, Kosari K, Humar A, Sielaff TD. Radiofrequency ablation causes ‘thermal fixation’ of hepatocellular carcinoma: A post-liver transplant histopathologic study. Clin Transplant 2003; 17: 377–384
- He X, McGee S, Coad JE, Schmidlin F, Iaizzo PA, Swanlund DJ, Kluge S, Rudie E, Bischof JC. Investigation of the thermal and tissue injury behaviour in microwave thermal therapy using a porcine kidney model. Int J Hyperthermia 2004; 20: 567–593
- Germer C, Isbert CM, Albrecht D, Ritz JP, Schilling A, Roggan A, Wolf KJ, Muller G, Bahr H. Laser-induced thermotherapy for the treatment of liver metastasis. Correlation of gadolinium-DTPA-enhanced MRI with histomorphologic findings to determine criteria for follow-up monitoring. Surg Endosc 1998; 12: 1317–1325
- Ozaki T, Tabuse K, Tsuji T, Nakamura Y, Kakudo K, Mori I. Microwave cell death: Enzyme histochemical evaluation for metastatic carcinoma of the liver. Pathol Int 2003; 53: 837–845
- Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am J Physiol Cell Physiol 2003; 284: C1309–C1318
- Allwork SP, Bentall HH. Usefulness of the phenomenon of histofluorescence in the identification of early myocardial necrosis. Cardiovasc Res 1986; 20: 451–457
- Anderson CD, Lin WC, Beckham J, Mahadevan-Jansen A, Buttemere CR, Pierce J, Nicoud IB, Wright Pinson C, Chari RS. Fluorescence spectroscopy accurately detects irreversible cell damage during hepatic radiofrequency ablation. Surgery 2004; 136: 524–531
- Chopra P, Sabherwal U. Histochemical and fluorescent techniques for detection of early myocardial ischemia following experimental coronary artery occlusion: A comparative and quantitative study. Angiology 1988; 39: 132–140
- Salinas-Madrigal L, Sotelo-Avila C. Morphologic diagnosis of acute tubular necrosis (ATN) by autofluorescence. Am J Kidney Dis 1986; 7: 84–87
- Wu BP, Tao Q, Lyle S. Autofluorescence in the stem cell region of the hair follicle bulge. J Investig Dermatol 2005; 124: 860–862
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995; 146: 3–15
- Majno G, La Gattuta M, Thompson TE. Cellular death and necrosis: Chemical, physical and morphologic changes in rat liver. Virchows Arch Pathol Anat Physiol Klin Med 1960; 333: 421–465
- Liang J, Wu WL, Liu ZH, Mei YJ, Cai RX, Shen P. Study the oxidative injury of yeast cells by NADH autofluorescence. Spectrochim Acta A Mol Biomol Spectrosc 2007; 67: 355–359
- Dragowska WH, Warburton C, Yapp DT, Minchinton AI, Hu Y, Waterhouse DN, Gelmon K, Sov K, Woo J, Masin D, et al. HER-2/neu overexpression increases the viable hypoxic cell population within solid tumors without causing changes in tumor vascularization. Mol Cancer Res 2004; 2: 606–619
- Shaked Y, Ciarrocchi A, Franco M, Lee CR, Man S, Cheung AM, Hicklin DJ, Chaplin D, Foster FS, Benezra R, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science 2006; 313: 1785–1787
- Shafirstein G, Hennings L, Kaufmann Y, Novak P, Moros EG, Ferguson S, Siegel E, Klimberg SV, Waner M, Spring P. Conductive interstitial thermal therapy (CITT) device evaluation in VX2 rabbit model. Technol Cancer Res Treat 2007; 6: 235–246
- Shafirstein G, Novak P, Moros EG, Siegel E, Hennings L, Kaufmann Y, Ferguson S, Myhill J, Swaney M, Spring P. Conductive interstitial thermal therapy device for surgical margin ablation: In vivo verification of a theoretical model. Int J Hyperthermia 2007; 23: 477–492
- Robinson DS, Parel JM, Denham DB, Gonzalez-Cirre X, Manns F, Milne PJ, Schachner RD, Herron AJ, Comander J, Hauptmann G. Interstitial laser hyperthermia model development for minimally invasive therapy of breast carcinoma. J Am Coll Surg 1998; 186: 284–292
- Robinson DS, Parel JM, Denham DB, Manns F, Gonzalez X, Schachner R, Herron A, Burdette EC. Stereotactic uses beyond core biopsy: Model development for minimally invasive treatment of breast cancer through interstitial laser hyperthermia. Am Surg 1996; 62: 117–118
- Wildhirt SM, Suzuki H, Horstman D, Weismuller S, Dudek RR, Akiyama K, Reichart B. Selective modulation of inducible nitric oxide synthase isozyme in myocardial infarction. Circulation 1997; 96: 1616–1623