
Abstract
FDA approval of anti-CTLA4 in 2011 for melanoma immunotherapy was paradigm shifting and dramatically accelerated cancer immunotherapy research. The investment and effort have been exceptionally large, with a commensurate impressive pace of discovery. Historical and current research has validated the following key points: tumors are recognized by the immune system; tumors develop an immunosuppressive environment which suppresses the antitumor immune response; successful immunotherapy must overcome that tumor-mediated immunosuppression. While cancer immunotherapy research expanded, a parallel effort developing nanoparticles (NP) for cancer diagnosis and therapy also received major investment and expanded. Initially the two efforts appeared to have minimal synergy. Systemically administered nanoparticles are rapidly ingested by phagocytic leukocytes, and therefore nanotechnologists developed strategies to avoid NP ingestion by leukocytes in order to accomplish nanoparticle accumulation in tumors rather than liver and spleen. Recently, nanotechnology and cancer immunotherapy have increasingly merged since phagocytic leukocytes are the key to reversing the local tumor immunosuppression and the tendency of NP to be phagocytosed can be exploited to manipulate phagocytes for immunotherapy. This review focuses on in situ vaccination (ISV), an immunotherapy approach that can utilize direct injection of immunostimulatory reagents, including NPs, into tumors to disrupt the local immunosuppression, stimulate effective immune response against the treated tumor, and most importantly, generate a systemic antitumor immune response to eliminate metastatic tumors. While there are many specific options for using NP for ISV (reviewed further in this special issue), this review focuses on immunology concepts needed to understand and design successful NP ISV approaches.
Cancer immunotherapy mechanisms and current barriers
As with many revolutions, scientific or not, a single event, in this case, FDA approval of the CTLA-4 blocking antibody Ipilimumab, was the visible manifestation for what had been developing for many years. As far back as 1890, there were organized scientific investigations in cancer immunotherapy by William Coley, including a variety of treatment efforts that injected bacteria either systemically or locally, including directly injected into tumors. The direct injection of bacteria into tumors is now recognizable as ISV [Citation1,Citation2]. Despite the work of Coley and others that followed, tumor immunology was a small, slow-growing area of research until FDA approval of anti-CTLA4 to treat metastatic melanoma in 2011 [Citation3,Citation4]. This FDA approval was a ‘shot heard round the world’ and along with subsequent approval of anti-programmed cell death protein/programmed death-ligand 1(PD-1/PD-L1) blocking antibodies, has tremendously expanded the immunotherapy-based treatment of cancer. Anti-CTLA4 and anti-PD1/PD-L1 are the main examples of a broader approach called various names, including ‘checkpoint blockade therapy’, (CBT), because they block suppressive signals (checkpoints) mediated through molecules like CTLA-4 and PD-1 expressed by many effector T cells [Citation5]. Reflecting this exploding research effort in cancer immunotherapy, there are 5706 papers in PubMed published in 2019 with both ‘cancer’ and ‘immunotherapy’ in the abstract, over 15 published per day on average.
The significance of immunotherapy lies in the fact that it is a new strategy to treat metastatic disease, which causes most cancer mortality. The dramatic potential of cancer immunotherapy has expanded interest beyond immunologists and attracted contributions from scientists trained in many disciplines, including chemistry, engineering and physics. It is anticipated that utilizing nanotechnology to develop novel immunotherapy approaches will advance cancer patient treatments. Reflecting that understanding, a large proportion of nanotechnologists developing cancer treatments have become focused on immunotherapy and are increasingly doing immunological assays in their labs or with collaborating immunologists. At the October 2019 NCI Alliance for Nanotechnology in Cancer PIs meeting, roughly 2/3 s of the presentations were focused on using nanotechnology to stimulate antitumor immunity to treat cancer. Likewise, at the 2019 annual Society of Thermal Medicine meeting, a variety of nanotechnologists and engineers reported on designing and utilizing nanoparticles specifically to stimulate antitumor immune responses against various tumor subtypes (PDF of the meeting agenda at https://www.thermaltherapy.org/ebusSFTM/ANNUALMEETING/2019ANNUALMEETING.aspx).
One aspect of cancer immunotherapy where nanotechnology stands to provide significant benefit is in reversing the immunosuppression within the tumor microenvironment (TME) (summarized by ). The immune system does recognize cancer as a threat and likely eliminates many tumors prior to clinical recognition [Citation6,Citation7]. However, the antitumor immune response is often weak and blocked by immunosuppression, and this allows some tumors to escape immune surveillance, develop and spread. The immune system evolved primarily to recognize pathogens and is quite good at that task [Citation8,Citation9]. Selection pressure to protect against pathogens has been very strong since historically, infectious diseases often eliminated children whose immune system was unable to protect against a given pathogen before they were able to have their own offspring [Citation10–12]. In contrast, the selection pressure imposed by cancer, a disease primarily of post-reproductive humans, has been much less than that of pathogens, suggesting less robust selection for tumor elimination by the immune system.
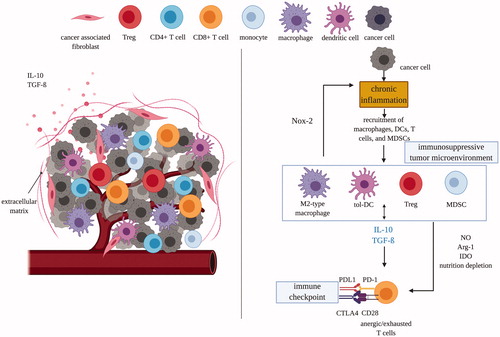
Figure 1. Tumor microenvironments are immunosuppressive which protects tumor cells and transforms immune cells from immunostimulatory to immunosuppressive. Left panel: tumor cells attract and recruit immune cells, such as macrophages, dendritic cells, and T cells. Right panel: however, the immunosuppressive nature of TME quickly suppress the activity of CD8 T-cells through multiple mechanisms mediated by MDSCs, tolerogenic DCs, M2-type macrophages and Treg cells. DC: dendritic cell; tol: tolerogenic; IDO: indoleamine 2,3-dioxygenase; Arg1: arginase; MDSC: myeloid-derived suppressor cells; NOX-2: nitric oxide synthase-2; PD(L)1: programmed death (ligand)-1; CTLA4: cytotoxic T lymphocyte-associated antigen; NO: nitric oxide; Treg: regulatory T-cell; IL: interleukin; TGF-β: transforming growth factor-β.
The adaptive arm of the immune system, B cells and T cells, recognizes ‘antigens’, which are usually ‘non-self’ molecules. While there is growing understanding that B cells and the antibodies they make play a significant role in resisting and eliminating tumors [Citation13,Citation14], immunologists consider T cells the most potent antitumor effector cells, and current cancer immunotherapy research is predominantly focused on T cells, (checkpoint blockade therapies specifically block suppressive signals to T cells). Cancer cells are ‘self’ with few recognizable antigens, unlike microbial pathogens, which have many obvious antigens and many characteristics that the immune system is literally hard-wired to recognize [Citation15,Citation16]. The question of whether and how cancer cells are recognized by the immune system has been discussed since the 1960s when understanding the mechanistic basis of the immune system accelerated, and experiments showed the existence of antigens that mediated the rejection of some tumors when transplanted between syngeneic hosts [Citation17–19]. This coincided with the recognition that viruses could cause cancer and the understanding that viruses carry antigens, so cancer that expresses viral genes is more easily recognized by the immune system [Citation20]. It is now accepted that tumors not virally infected also express recognizable antigens, but these are often not very strong immune targets since they are always at least similar to normal proteins that the immune system has been tolerized to, that is, trained to not respond against. Antigen variability is an important issue that impacts antitumor immune responses. Tumors, both by type and within a type between patients, are highly variable in the antigens they express, making some tumors quite recognizable (immunogenic or ‘hot’) and others very weakly recognized (nonimmunogenic or ‘cold’) [Citation21,Citation22].
Antigens in tumors are of 3 general classes, tumor-associated antigens (TAAs), tumor virus antigens, and neoantigens. For many years, tumor immunologists focused on TAAs or viral antigens since they were technically accessible to study (example of TAA is NY-ESO-1 or mesothelin, examples of viral antigens are human papillomavirus E6 or E7). TAAs are normal human proteins often expressed in some way abnormally, on the wrong tissue, at the wrong time during development, or at a level much higher than normal. One obvious downside of TAAs is that the immune response against them is generally weak. Since these are normal proteins, the immune system has seen them before as ‘self’ and has been ‘tolerized’ to not respond to them as antigens. However, we know from autoimmunity that normal proteins can be treated as antigens and TAAs are valid targets for immunotherapy but, to have efficacy, treatments dependent on TAAs must overcome some level of immune tolerance. The second downside of TAAs is that, since they are normal proteins, they are often also expressed by other cells in the patient, and an immune response against a TAA can lead to autoimmunity. An interesting positive example of response against TAA causing autoimmunity is the development of vitiligo in melanoma patients. Vitiligo patients have patches of skin that are very pale due to the elimination of melanocytes by the immune system. The immune system attacking the melanoma cancer cells also recognizes TAA found in normal melanocytes, leading to their elimination. Vitiligo in melanoma patients is considered a positive sign that the immune system recognizes and is attacking the melanoma [Citation23]. While the elimination of skin melanocytes by autoimmunity is not life-threatening, the autoimmune response against normal cells in vital organs like lung or intestine that are common cancer sites could cause intolerable side effects.
The second type of recognizable antigens that are found in most tumors are ‘neoantigens’, that is new antigens [Citation24]. These novel antigens are due to mutations in tumor cells, and many tumors carry dozens, hundreds or even thousands of mutations [Citation25]. With the advent of whole genome sequencing, patient-specific neoantigens in tumors are now identifiable and are the focus of therapeutic vaccines that include peptide targets from neoantigens [Citation26,Citation27]. T cells respond to short peptides presented by human leukocyte antigen (HLA) complexes on antigen-presenting cells as antigens (generally 8-11 amino acids for class I and 13-17 for class II) [Citation28]. A single point mutation may generate a strongly recognizable new antigen, although, due to the biology of antigen presentation, most mutations do not generate a strong antigen [Citation27,Citation29]. Whether they will be recognized, and how strongly, depends on a variety of variables that includes the specific mutation, whether that region of the protein is efficiently processed into a peptide and whether that peptide is efficiently presented to T cells by an allele of the patient's highly variable HLA (class I and class II) [Citation28,Citation30]. So, the more mutations that a tumor carries, the more likely that there will be a greater number of recognizable neoantigens. The correlation between mutation number and response to checkpoint blockade therapy (CBT) has been statistically demonstrated, and tumor types and patients with more mutations are more likely to respond to CBT [Citation22,Citation31,Citation32]. However, although the potential for well-recognized neoantigens increases with the number of mutations, it is a probability, and tumors with very few mutations could express strong neoantigens and respond well to CBT, while tumors with a lot of mutations may not. It is important to note that unlike TAAs, there is little or no immune tolerance to many neoantigens and so response against neoantigens are likely to be stronger than against TAAs. This raises a challenge for T cell-mediated cancer immunotherapy. TAA found on a given type of tumor may apply to a large proportion of that tumor type, making an immunotherapy strategy to target a TAA usable for many patients. However, since the vast majority of neoantigens in tumors are unique to a given patient, treatment focused on identifying and vaccinating against neoantigens is truly personal therapy and must be customized for each patient. As discussed below, one strength of the ISV approach is that there is no need to identify or specifically utilize TAAs neoantigens or viral antigens. The tumor is the source of the antigens, regardless of their type, and understanding the available antigens intellectually does not impact the therapeutic approach of ISV.
Beyond presence or absence of well-recognized antigens, the other major barrier to effective antitumor immune responses is the immunosuppressive tumor microenvironment (TME). Immune responses activate and deploy an army of ‘trained killers’, which like any situation with lethal power, needs to be controlled. There are many layers of control systems to protect the host from autoimmunity, and a major one is that immune cells respond to suppressing as well as stimulating signals. As soon as an immune response is generated, the signals that limit it and prevent autoimmunity are also activated. There is an evolutionary balance between damage due to pathogens and damage due to autoimmunity, both of which can lead to reduced reproduction for an individual [Citation33]. Even when the immune system strongly recognizes the tumor antigens and responds, the effector cells recruited to the tumor receive a variety of immunosuppressive signals from the tumor and from leukocytes the tumors recruit. This blocks the effector response and limits efficacy of immunotherapy through a variety of cells and mechanisms [Citation34]. For example, M2 macrophages generate suppressive cytokines, such as interleukin 10 (IL-10) and transforming growth factor-β (TGF-β), to suppress the activity of cytotoxic CD8+ T cells [Citation35,Citation36]. Overall the tumor sets up a local environment that supports recruitment and proliferation of immunosuppressive cells and modifies other cells to have an immunosuppressive function like regulatory T cells (T regs) [Citation37,Citation38], tolerogenic dendritic cells (tol-DCs) [Citation39,Citation40], and myeloid-derived suppressor cells (MDSCs) [Citation41–43]. This immunosuppressive local tumor environment suppresses adaptive immune responses against tumors and thereby enables cancer to take hold and spread () [Citation44].
CBT for CTLA-4 and PD-1/PD-L1, the current widely used immunotherapies, work on the problem of tumor-generated immunosuppression by blocking specific signals that suppress T cell responses, through surface receptors on T cells, CTLA-4 and PD-1. CBT is administered systemically, and the antibodies are at high levels all over the body. However, checkpoint blockade therapy is not specifically targeting T cells that recognize tumor antigens, CBT is not ‘antigen-specific’. If we consider the checkpoint molecules on the surface of T cells as ‘brakes’ on their response, systemic administration of CBT is taking the brakes off all the T cells, not just those that recognize the tumors. Not surprisingly, CBT often leads to therapy-limiting autoimmunity as other T cells that recognize ‘self’ lose their inhibitory signals and damage normal tissues [Citation45–47].
What is in situ vaccination (ISV) for cancer immunotherapy?
CBT immunotherapy to treat metastatic disease is administered systemically, much like chemotherapy. As with other systemic anti-cancer treatments, there are side effects, that limit their usage and efficacy, in this case, autoimmunity. The high frequency of therapy-limiting autoimmunity side effects when using a checkpoint blockade illustrates the challenge clearly [Citation45–47]. There are, however, many vaccine strategies that could be combined with CBT to generate improved results with potentially less autoimmunity by focusing the immune response against the tumor rather than against normal tissues [Citation48–51]. One such strategy is in situ vaccination (ISV) to support what is termed the cancer immunity cycle () [Citation52]. In ISV therapy, an immune-stimulatory reagent is injected or applied directly into one or a few tumors in a patient, causing the reversal of the tumor-mediated local immunosuppression, and optimally, generating a large number of antitumor T cells that effectively disperse through the body to attack cancer cells in metastases that may or may not be clinically recognized [Citation53,Citation54].
Figure 2. In situ vaccination can modulate tumor microenvironments to induce anti-tumor immune responses at multiple levels in the cancer immunity cycle. (A) ISV with immune adjuvant can cause local tissue damage, inducing immunogenic cell death and release of cancer cell antigens; and/or directly introduce PAMPs and/or DAMPs with TLR agonists or NPs into the tumor to (B) attract and activate dendritic cells and other antigen-presenting cells, (C) which mature and migrate from primary tumor to stimulate the priming and activation process of T cells in the tumor-draining lymph nodes. (D) Once primed and activated, T cells will then traffick to and (E) infiltrate solid tumors and metastases to (F) directly kill the tumors with granzymes and perforins. Multiple ISV strategies can be combined to achieve stronger anti-tumor immune response. ICD: immunogenic cell death. TAAs: tumor associated antigens. Immune adjuvants: injection of immune stimulating reagents. DAMPs: danger-associated molecular pattern molecules. PAMPs: pathogen associated molecular pattern molecules. HIFU: high intensity focused ultrasound. TLR: toll like receptor. NP: nanoparticles. Adapted from Chen and Mellman [Citation52].
![Figure 2. In situ vaccination can modulate tumor microenvironments to induce anti-tumor immune responses at multiple levels in the cancer immunity cycle. (A) ISV with immune adjuvant can cause local tissue damage, inducing immunogenic cell death and release of cancer cell antigens; and/or directly introduce PAMPs and/or DAMPs with TLR agonists or NPs into the tumor to (B) attract and activate dendritic cells and other antigen-presenting cells, (C) which mature and migrate from primary tumor to stimulate the priming and activation process of T cells in the tumor-draining lymph nodes. (D) Once primed and activated, T cells will then traffick to and (E) infiltrate solid tumors and metastases to (F) directly kill the tumors with granzymes and perforins. Multiple ISV strategies can be combined to achieve stronger anti-tumor immune response. ICD: immunogenic cell death. TAAs: tumor associated antigens. Immune adjuvants: injection of immune stimulating reagents. DAMPs: danger-associated molecular pattern molecules. PAMPs: pathogen associated molecular pattern molecules. HIFU: high intensity focused ultrasound. TLR: toll like receptor. NP: nanoparticles. Adapted from Chen and Mellman [Citation52].](/cms/asset/e4a5d61f-f02b-49ef-9150-301ced198a86/ihyt_a_1810333_f0002_c.jpg)
We define ISV to have the following characteristics: ISV is directly applied to one or more recognized tumors with the express goal of generating local and systemic antitumor immune response; while ISV could include treatments that cause immunogenic cell death that has a strong effect on the treated tumor, (ICD, described more in detail below and in ), the systemic effects are mediated by the immune system rather than direct cell death caused by treatment; ISV does not bring in tumor antigens of any sort, but rather depends on the antigens within the tumor as the antigen source for the vaccine.
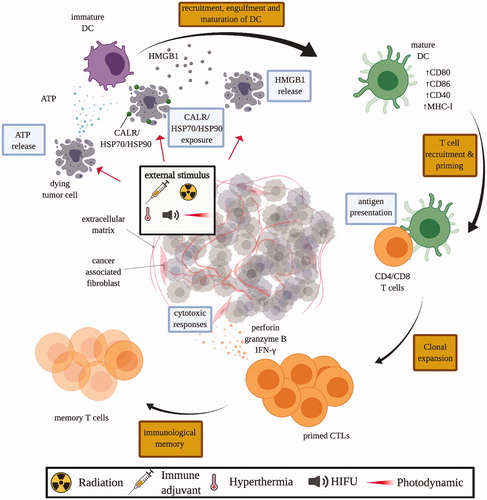
Figure 3. Mechanisms of immunogenic cell death (ICD). Tumor cells subjected to ICD-inducing treatment, such as chemotherapeutics, immune adjuvant injection, radiation therapy, HIFU, and photodynamic therapy, release damage-associated molecular pattern (DAMP) molecules like calreticulin (CALR), adenosine triphosphate (ATP), heat shock protein 70 and 90 (HSP70/90), and high mobility group box 1 (HMGB1). These DAMP molecules stimulate the recruitment and maturation of dendritic cells (DCs), which facilitates the engulfment of tumor antigens, and antigen presentation to T cells in the draining lymph nodes. Consequently, matured DCs trigger the priming of an adaptive immune response against tumor cells by T cells. Primed cytotoxic T lymphocytes (CTLs) expand and leave the lymph nodes that drain the treated tumors, circulate and identify other tumors by antigen expression, and kill untreated tumors with cytoxic effector molecules like perforin-1 and granzyme B.
The ISV approach to cancer immunotherapy was first investigated and practiced by Coley, although he was not using the term or aware of the specific concepts. Coley was a cancer surgeon and knew of patient cases in which an infection at the surgical site following removal of one or more tumors was followed by shrinkage of tumors that were not removed. He tried to reproduce this as therapy and reported that injection of bacteria into one or more tumors in patients with multi-tumor metastatic disease was sometimes followed by shrinkage of untreated tumors [Citation2]. This was reported in a number of publications and, although remarkable when it occurred, was sporadic and poorly reproducible in other people’s hands, and Coley’s approach did not gain widespread application [Citation55].
At roughly the same time as Coley was experimenting with bacteria to treat cancer, radiation was coming into widespread use to treat tumors locally. It took almost roughly 80 years for it to be recognized that radiation can be part of cancer immunotherapy. Essentially, radiation (RT) can be a mediator of in situ vaccination as can other physical tumor-damaging treatments like hyperthermia, high intensity focused ultrasound (HIFU), or photodynamic therapy (PDT) [Citation56–63]. The term ‘abscopal effect’ in regard to radiation was first used in a 1953 report that documented physiological effects away from a part of the body exposed to radiation, but this was not referring to cancer [Citation64]. By the late 1960s, case reports appeared in which irradiation of some tumors was followed by shrinkage of tumors that had not been irradiated, and ‘abscopal effect’ was used to refer to these increasingly reported results and is the common term for this effect in radiation oncology [Citation65–67]. Perhaps surprisingly, it was not until 2004 when it was proposed that the radiation abscopal effect against tumors was immune-mediated [Citation68]. While still very uncommon, many more reports of the RT abscopal effect have been made since then, and it has been reproduced in animal models so that now it is recognized as the generation of a systemic antitumor immune response by local tumor radiation treatment [Citation69]. Currently, it is an important topic in radiation oncology research and designing RT to primarily generate the abscopal effect is ISV, since it fits the definition given above. The rare occurrence of a detectable abscopal effect from RT alone suggests that, like most cancer immunotherapies, it is likely to be one part of a multimodal combinatorial immunotherapy strategy. While first applied to RT, the term abscopal effect is applicable to other forms of ISV, including the application of other energy-depositing treatments like hyperthermia, HIFU, and PDT.
From an immunological perspective, the mechanism of in situ vaccination with strongly immunostimulatory reagents (immune adjuvants), like live, attenuated, or killed bacteria, is explainable as local immune stimulation that causes a reversal of local immunosuppression and activates systemic immune response against untreated tumors. However, the immune mechanism of the RT abscopal effect has been more difficult to determine, and perhaps this is why it was not quickly recognized. One reason for this slow appreciation of the immune basis of the abscopal effect is because radiation, in general, is immunosuppressive due to the hypersensitivity of leukocytes to radiation. Over the last 20 years, a new immunologic concept emerged that explains the RT abscopal effect by what is now known as ‘immunogenic cell death’ (ICD, ) [Citation70]. ICD was first identified as an immunostimulatory form of cell death that certain chemotherapeutic cancer drugs generate as part of their efficacy [Citation71]. ICD has a number of hallmarks that are immunostimulatory, and the ones most often assayed to verify ICD are the release of ATP, the release of the nuclear protein high mobility group box 1 (HMGB1), and binding of calreticulin, an internal chaperone protein, on the external surface of the dying cells [Citation72]. Of interest for thermal medicine, the release of heat shock proteins is also an immunogenic signal of ICD [Citation73–75]. The tendency of RT to generate ICD is now accepted as the immunologic basis of the RT abscopal effect. However, as noted, RT by itself rarely generates effective systemic immune response [Citation76]. Although direct tumor-killing is accepted as the primary mechanism by which RT eliminates tumors, it is an open question regarding how much total elimination of irradiated tumors involves a local immune response.
Generating ICD of tumor cells is clearly of value for in situ vaccination. Recently we (AR lab) tested whether generating ICD signals to the immune system without an increase in immunogenic cell death directly is of value for ISV. The study injected lipid nanoparticles carrying calreticulin expression plasmids into mouse tumors to test whether calreticulin overexpression could generate a signal similar to external cell membrane-bound calreticulin due to ICD. It worked well by itself in reducing treated tumor volume and was improved when combined with moderate high intensity focused ultrasound which would increase tumor cell death, ICD, and antigen release [Citation77]. This provides a new conceptual approach to supporting ISV by artificially generating the immune signals of immunogenic cell death. Using nanoparticles to cause or mimic ICD could increase the systemic efficacy and dependability of in situ vaccination strategies.
The above ideas all help to understand the current status of in situ vaccination and why it is one focus of cancer nanotechnology efforts. However, the question of what makes this a ‘vaccination’ has not been discussed and it is relevant. All vaccines carry 2 functional components, regardless of their ingredients. One is an antigen, the molecule(s) that the immune system is being trained to recognize and respond against. The other is ‘immune adjuvant’, a term denoting a reagent that stimulates the immune system to recognize and respond against the antigen, rather than ignoring it or tolerizing to it. Immune adjuvants are essentially dangerous signals that tell the innate immune system cells that there is a pathogen that requires an immune response against associated antigens. All vaccines, with the exception of in situ vaccines, carry both antigen and adjuvant. In situ vaccination does not carry antigens but is solely adjuvant (including ICD) applied directly to tumors. This raises the question of what ISV uses as antigen, and the answer is that the tumor itself is the source of the antigens. As noted previously, tumors carry tumor-associated and neoantigens, and one of the advantages of ISV, as compared to many other therapeutic cancer vaccination strategies, is that there is no need to identify what antigens are in the tumor and which are most recognizable by the immune system. All relevant antigens exist in the tumor; if they are not in the tumor, they are not relevant for immunotherapy of any sort. By putting the adjuvant in the tumor, ISV combines the antigen in the tumor cells with the adjuvant and creates the vaccine. This ISV approach does not negate the potential of other approaches that utilize TAAs in vaccines or identify potential neoantigens and predict what neoantigen will be best presented on class I or II HLA of each specific patient by antigen-presenting cells, so that peptide can be used in a vaccine [Citation78–80]. Immunotherapy is increasingly understood to need a variety of combinatorial approaches to overcome the inherent difficulties in any cancer therapy, and ISV is certainly going to contribute to those combinations, as will approaches that identify and vaccinate with neoantigens and/or tumor-associated antigens. provides a graphical summary of in situ vaccination.
Why utilize in situ vaccination to treat identified tumors when most can be eliminated with surgery or radiation?
This question addresses the central concept of in situ vaccination. When involved in cancer research and therapy development, the readership of IJH is predominantly focused on physically-based local treatments of tumors. For any novel local tumor treatments, there is a fundamental question that has to be answered; why is the approach better than surgery and/or radiation? There are specific situations that do not involve immunology in which something other than surgery or radiation is the current standard of care, such as using heat to eliminate tumors locally in the liver where surgical removal is quite often not possible due to tumor location [Citation81–83]. However, as a broad strategy for treating solid tumors, physically-based approaches outside of RT, such as hyperthermia, photodynamic therapy, or high intensity focused ultrasound are likely to be used only in special circumstances if the goal is the simple removal of a recognized tumor. The concepts of in situ vaccination and the associated immune-based abscopal effect provide the rationale for physical treatment of local tumors that are designed to boost systemic antitumor immune responses in order to fight established or suspected metastatic disease, which surgery does not do at all and RT does poorly by itself [Citation84–86].
Though currently applied to a limited range of indications, ISV treatments are used clinically. ISV has been in use clinically for superficial bladder cancer since 1977 [Citation87]. Superficial bladder cancer is a very serious diagnosis, despite the descriptor ‘superficial’, and is often metastatic. The standard of care is surgical tumor removal followed by repeated instillation into the bladder of attenuated cow tuberculosis, Bacillus Calmette-Guerin (BCG), that is widely used to vaccinate against tuberculosis (TB). Essentially this is modeling a surgical site infection following tumor surgery. Interest in BCG for cancer treatment began when a 1959 Nature paper reported that BCG application could protect mice from transplanted tumors [Citation88]. BCG does not carry any antigen found in the tumor; it is used as an adjuvant to stimulate the immune system; it is ISV when used to treat human bladder cancer. Clinical trials initiated in the 1970s compared BCG in situ vaccination with then-current chemotherapy drugs to treat bladder cancer patients without known metastatic disease, and BCG was superior at preventing metastatic disease development [Citation88–91]. This later became the first FDA approved cancer immunotherapy and is still the standard of care for superficial bladder cancer despite significant progress in chemotherapy in general [Citation92].
Another recent example of FDA approved ISV is T-Vec (Imlygic, Amgen Inc.), a genetically modified herpes simplex type I virus. Part of a broad strategy of cancer therapy, oncolytic viruses (OVs), it was approved in late 2015. OVs can lyse cells and be genetically modified to preferentially lyse tumor cells rather than normal cells. The original and very attractive idea behind oncolytic viruses was that they could be systemically administered and since they preferentially replicate in tumors, would infect, replicate and kill tumor cells in an immunostimulatory way. So far, this systemic administration strategy has not been supported by clinical trials, but this approach may still have merit since relatively few viruses have undergone clinical trials and the engineering space is quite open. In contrast to the failure of systemic administration, T-Vec injected directly into melanoma on the skin was beneficial and that is the current FDA approved usage [Citation93]. T-Vec is engineered to recruit antigen-presenting cells by making GMCSF, which further supports antitumor immune responses and illustrates another ISV strategy of producing cytokines in the tumor. T-Vec is used clinically by injection into tumors to stimulate antitumor immune responses (adjuvant), causes immunogenic cell death, and does not carry tumor antigens [Citation94]. It is worth noting that almost all viruses are in the size range of nanoparticles so when we consider nanoparticles for cancer immunotherapy, engineered oncolytic viruses are NPs. Therefore T-Vec used this way is using NP for in situ vaccination. Another example of increasing FDA approval of ISV strategy was the recent approvals of rose Bengal dye (PV-10) for intratumoral injection in melanoma and neuroblastoma in 2018. While directly mildly cytolytic, the drug stimulates antitumor immunity by inducing ICD and in mice and patients, regression of uninjected tumors occurs, demonstrating systemic immune responses [Citation95–98].
The intratumoral injection is a fundamental aspect of most ISV and can be criticized as a limitation when it comes to treating tumors that are not superficial. It is true that near-surface tumors, such as skin or breast tumors, are easier to inject, however using imaging modalities like ultrasound or others, surgeons and interventional radiologists can safely inject NPs into almost any anatomic site. Thus, although some tumors will be easier to access than others, ISV treatment is not limited to a narrow subset of anatomical locations.
Why do nanoparticles have unique potential for in situ vaccination?
The first wave of nanotechnology strategies to treat cancer focused on systemically applying NP and expecting them to concentrate in the tumor preferentially due to what was termed ‘enhanced permeability and retention’, known as the EPR effect to prepare the circulating immune cells to identify and eliminate cancer cells [Citation99,Citation100]. EPR describes a sporadic characteristic of tumor vasculature that allows systemically delivered NP to accumulate preferentially in tumors [Citation101,Citation102]. EPR has been successfully exploited for reducing chemotherapy toxicities clinically (e.g., Doxil), but it has also been found that the EPR-based NP delivery approach for delivering chemotherapy is not superior to conventional therapies in terms of enhancing survival rates [Citation103,Citation104]. This is explained in part by the minimal EPR effects in most human tumors and the heterogeneous distribution of NP with EPR into tumors that do exhibit EPR [Citation105].
As reagents for cancer therapy, NP provides a number of advantages including but not limited to: an improved platform for drug delivery [Citation106,Citation107], which can overcome the low bioavailability of administered reagents [Citation108] including exploiting the tendency of phagocytes to ingest NP to manipulate antigen-presenting cells [Citation109]. A wide variety of nanoparticles have been studied, although generally by systemic administration rather than intratumorally, so not ISV. One way to categorize these NP is based on the material: organic, inorganic, lipid, glycan nanoparticles as well as synthetic polymers [Citation110,Citation111]. Examples include gold NP [Citation112], iron oxide NP [Citation113], solid lipid NP [Citation114], plant viral particle [Citation115] and nanoscale coordination polymer NP [Citation116]. A more detailed review of NP studied with ISV can be found in Gorbet et al., in this special issue.
One aspect of the systemic administration of NP for cancer therapy is that NP-size particles are aggressively phagocytosed by phagocytic leukocytes. Phagocytes are typically the first line of responders, which are also in charge of priming adaptive immune cells, such as CD4+ and CD8+ T cells [Citation117]. Most NPs administered parenterally are phagocytosed by leukocytes in the liver and spleen. Efforts to hide the NP from leukocytes with various coatings, most often polyethylene glycol (PEG), have enhanced circulatory half-life, but this strategy did not significantly increase the accumulation of intravenous NP in tumors in humans [Citation118]. Alternative approaches to put ‘targeting’ molecules on NP were developed in which antibodies or receptor ligands that bind to surface proteins on tumors were attached to the NP. This enabled the low proportion of NP that did get into the tumor to more frequently adhere to the tumors and stay there, that is, to enhance the ‘retention’ of these NPs [Citation119–121]. However, although efforts continue, to date getting systemically administered NP to preferentially accumulate in tumors has not had consistent success in animal models or humans [Citation122]. By using NP as ISV reagents directly injected into tumors, this major challenge of NP drug delivery is avoided, although at the cost of not directly delivering NP to untreated tumors.
In the context of immunotherapy, there is an extraordinary opportunity for NP utilization. The tendency of NP to be phagocytosed by leukocytes creates an efficient route to manipulate the immunosuppressive phagocytes that are found in tumors. Injection of NP into tumors concentrates the NP in the phagocytes in the tumor and draining lymph nodes and supports the immunologic targeting of those crucial immunosuppressive cells [Citation123,Citation124]. Phagocytic leukocytes are the forward sentinels of the immune response, their response determines whether adaptive immune cells, the lymphocytes, will respond or not. The immune control mediated by phagocytes involves both stimulation and suppression, and they are major mediators of the immunosuppression by tumors. While the existence of immunosuppressive immune cells seems to be a paradox, it is clear that virtually every type of leukocyte has both immunostimulatory and immunosuppressive functions [Citation125,Citation126], and the immune outcome depends on the balance of these opposing functions which is controlled by an array of regulatory inputs that each cell experiences. Accumulation of NP in immunosuppressive phagocytes within tumors provides a powerful mechanism to manipulate these immune cells and stimulate antitumor immunity [Citation113,Citation124,Citation127]. Phagocytes, in general, can move quickly from immune suppression to immune stimulation or vice versa depending on environmental signals. One consequence of effective ISV is modulating the phagocytic leukocytes within the tumors so that they are no longer immunosuppressive and instead become effective immunostimulatory and antigen-presenting cells to elicit strong antitumor effects [Citation128–130]. Intratumoral injection eliminates the challenge of getting the NP to the tumor and ISV using NP provides a powerful opportunity to manipulate intratumoral phagocytes. Another useful but not yet developed potential for ISV with NP is that NP can be multicomponent. This has to be carefully studied and it has not yet been. An example of the complexity is a study in which intratumoral injection of free doxorubicin in addition to immunostimulatory plant viruses had better antitumor efficacy than attaching the drug directly to the virus [Citation129,Citation131]. While the reason for the difference is not fully elucidated, it is likely that attaching the drug to the NP delivers large amounts of drug to leukocytes and much less drug to tumor cells which would impair the antitumor immune response in multiple ways.
Combining PAMPs and DAMPs for ISV
ISV delivers or generates immunostimulatory molecules in recognized tumors. A critical question in ISV therapy is what reagents and treatment strategies generate the best local, and particularly the best systemic, antitumor immune responses. As noted above, there are 2 independent but related approaches that have value in ISV efforts. One is the adjuvant that stimulates the innate immune cells to respond as if the tumor is infected, and the other is the immunogenic cell death that can be generated by various tumor treatments and stimulates innate immune cells through different pathways. It is likely that having strong adjuvant as well as strong immunogenic cell death in ISV will lead to the best local and the strongest systemic antitumor effects because they work through different but complementary signaling pathways. However, that has not been clearly established.
Immunologists now recognize 2 basic classes of immunostimulatory molecules, pathogen-associated molecular patterns (PAMPs, used as immune adjuvants) and danger-associated molecular patterns (DAMPs, generated by ICD). As the name implies PAMP-based molecules do not generally interact with cells unless there are microbes present. PAMP-molecules are recognized by many receptors, of which the largest and best-known group are toll-like receptors (TLRs) [Citation132]. Examples of promising use of TLR stimulation for ISV include the use of Poly-IC, a TLR3 agonist, to treat brain cancer and facial rhabomyosarcoma [Citation133–135]. In contrast, DAMPs are generated by immunogenic cell death or something that mimics ICD [Citation75]. We propose that delivering adjuvant in the form of PAMPs and concurrent induction of DAMPs will generate the best ISV treatment efficacy. In this regard, radiation, hyperthermia, photodynamic therapy, HIFU and other physical treatment modalities of interest to International Journal of Hyperthermia readers can play an important role through DAMP induction to help generate therapeutic abscopal effects. However, the questions involved in combining DAMPs and PAMPs for cancer immunotherapy have not been well studied to date. In part, this is because in many cases they are difficult to separate since a strong adjuvant can generate immunogenic cell death of tumor cells through stimulation of immune cell attack.
The role of ISV in combinatorial cancer immunotherapy
Aggressive cancers are typically treated with multiple modalities that combine surgery, chemotherapy, and radiation therapy, and now increasingly immunotherapy. Combinatorial treatment also applies specifically to immunotherapies in which the goal of increasing responses across tumor types mandates combinations of immunotherapies [Citation49]. Challenges of timing and dosing greatly complicate combinatorial treatments, especially for immunotherapy that depends on choreographed responses from an array of cell types which are also impacted by the therapies themselves. Since they often involve rapidly dividing cells, immune responses are sensitive to anticancer therapies that target dividing cells. An example of this challenge is how to combine radiation therapy with the injection of PAMP reagents for ISV [Citation50]. Radiation is usually administered in multiple doses. How would the multiple RT doses interact with the leukocytes drawn into the tumor by ISV administration of PAMPs or radiation generation of DAMPS? How long following a specific radiation dose is the release of DAMPs beginning, maximal and fading? What timing of DAMP and PAMP signaling generates optimal systemic antitumor immune response? Beyond the obvious issues of what approaches to use and how to dose them, there are many variables of timing to understand and it warrants extensive intellectual and animal modeling to get the best information out of research investments. Similar questions of timing apply to hyperthermia, PDT, HIFU or other energy deposition-based strategies that can generate DAMPs when the strategy includes application of PAMPs for ISV. Although this is likely to be less complex because these modalities are not often applied many times like radiation.
Significantly better outcomes from immunotherapies will require a deeper understanding of tumor immunology, developing new immunotherapy approaches, identifying the best combinations of multiple immunotherapies, and integrating multiple immunotherapies with other modalities. Immunotherapies of the future are likely to integrate systemic immune treatments like checkpoint blockade, local treatments like in situ vaccination, adoptive cell therapies, as well as new approaches to immune manipulation such as modifying the patient’s microbiome [Citation136,Citation137].
Combinatorial therapy possibilities increase exponentially with increasing options. To make rapid progress, researchers should base the treatment sequence and frequency on a rational understanding of the immunological characteristics of effective versus ineffective therapies in different clinical situations and understand what is limiting those that are not effective. Currently, there is a large and growing number of publications in which various strategies are combined with checkpoint blockade therapy since that is the leading approach, including the use of ISV with CBT [Citation134,Citation138,Citation139]. Combining CBT with ISV has the needed mechanistic immunologic basis for expectation of value. Checkpoint blockade therapy depends on the presence of T cells that recognize the tumors and are not unresponsive due to ‘exhaustion’ or immunosuppressive signals, and if the numbers of such cells are insufficient, it is one of many reasons why CBT fails. ISV can generate increased numbers of the tumor-recognizing effector T cells needed for CBT response and a number of publications clearly show the synergy between CBT and ISV in mouse models [Citation140]. While not clearly proven, it is likely that combining CBT with ISV will generally improve antitumor efficacy with less autoimmunity since, if ISV increases antitumor effector T cell numbers, then the response to CBT could be both faster and more effective, with potentially shorter CBT treatment and/or reduced dosages. As noted, more detailed preclinical and clinical studies are needed to understand the impact of combining ISV and CBT. As with all cancer therapies, the details of the biology of cancer types and in particular the specific reason that CBT may or may not work in a given patient is likely to make outcomes of combining CBT with the ISV variable. Importantly, toxicity from ISV appears confined to local inflammation, so it is likely to be safe and tolerable.
Specifically, ISVs that combine stimulation of PAMP receptors with ICD that releases DAMPs may produce optimal therapeutic outcomes. Since the PAMPs will have to be introduced from the outside, they are likely going to be injected reagents. The DAMPs on the other hand can come from the tumor cell death and could be generated in many different ways, including a variety of physics-based approaches such as radiation, heat, cold, high intensity focused ultrasound or phototherapy. Nanoparticles could be valuable for either PAMP or DAMP stimulation in a variety of different ways. The potential multi-reagent character of NPs gives them inherent capabilities to stimulate multiple pathways. Further, multiple NP reagents could be administered as part of ISV either together or sequentially. The challenge is to realize that most of the NPs injected into a tumor will be in the phagocytic leukocytes in the tumor or draining lymph nodes, not the tumor cells, and therefore the strategies must work with that reality.
Potential for ISV prior to surgery on a primary tumor (neoadjuvant ISV immunotherapy)
One opportunity with exceptional therapy potential is performing ISV on a primary tumor before surgery to eliminate the tumor. The standard pattern of clinical trials for new cancer therapies is testing in late-stage patients with extensive metastasis that have failed all standard therapies. The understandable and important reasons why therapies are not studied in earlier-stage patients are concerns about safety and unwillingness to disrupt the established standard of care. Most of these subjects are not only late stage but have failed multiple other therapies and of course, they are the most challenging patient population to treat, particularly for immunotherapy since large tumor burdens are considerably more immunosuppressive locally and systemically and considerably harder for the immune system to impact. The reality is that every cancer therapy is likely to be more effective in earlier-stage patients, but this is not how they are tested. We propose that ISV can elegantly fill the gap in pre-surgery immunotherapy for a large number of early-stage patients with pathologic diagnosis that indicates a high probability for metastasis. The local administration of ISV is inherently safer than systemic immunotherapy since locally high concentrations of reagents put into a tumor are low compared to what is currently delivered systemically. For example, systemic cytokine administration can powerfully stimulate an antitumor immune response, but clinical trials with systemic administration of IL-2 and IL-12 show that they cause severe and often intolerable side effects [Citation141–143]. While high local cytokine concentrations in a tumor will cause local inflammation, the overall systemic levels will be much lower and therefore safer and more tolerable [Citation134,Citation144,Citation145]. Researchers investigating immunotherapy for early-stage patients should keep in mind that any cancer treatment being done on early-stage patients must not interfere with the standard of care approaches.
Would ISV prior to surgery have benefit? It is completely feasible to effectively deliver ISV in the 1-3-week average time between pathologic diagnosis and surgery for most cancers. Stimulating the cancer immunity cycle is the basis of ISV (). Simply put, ISV changes the tumor microenvironment, increases antigen presentation of tumor antigens and generates immunogenic cell death to release tumor antigens and DAMPs. The newly activated antigen-presenting cells take up the antigen and, as part of the activation, leave the tumor and travel to zdraining lymph nodes, where they present antigen to T cells and stimulate naïve T cells to replicate, become effector cells and leave the lymph node in search of antigen-expressing cells, which they attack. Importantly, this whole cycle takes roughly a week and the portion that occurs within the tumor takes roughly 3 days, which means that ISV could be effectively done prior to surgical tumor removal [Citation84,Citation146]. The implications are that following pathologic diagnosis that documents a significant risk of metastatic disease, ISV using established and not patient-specific reagents could quickly be initiated so that local and systemic immunotherapy is already stimulated prior to surgical tumor removal. In effect, ISV could be done as neoadjuvant therapy for many patients, with no interference with the standard of care. This could provide initial immunotherapy in a safe manner for patients who are at risk of metastatic disease prior to such disease manifesting and when the metastatic disease is small and more vulnerable to immune attack.
Summary, future challenges and opportunities
ISV is now poised to become an integral part of multimodal cancer immunotherapy for patients with all stages of the disease. This is occurring more than 100 years after William Coley first performed and reported a variety of immunotherapy approaches, including ISV (without the ISV name). The advantages of ISV include local immune-mediated efficacy, systemic immune impact, simplicity, speed of delivery, safety, modest cost, and ability to combine with other immunotherapies and with the established modalities of radiation, chemotherapy and surgery. All of these characteristics support the expectation of clinical utility and widespread application. In this context, nanoparticles are particularly promising because they are phagocytosed by immunosuppressive leukocytes in tumors, which provides an opportunity to manipulate these cells for the generation of an immune-competent TME and expanded tumor-specific effector T cell numbers. Many challenges must be addressed, particularly identifying the best ISV strategies for each tumor type and the best reagents for that strategy, and then integrating these approaches with other immunotherapies and with other modalities. There are many variables to consider and each one needs to be isolated and studied in preclinical models and applied and evaluated in patients. One important capability of nanotechnology is making NP carrying multiple different components with potential impact in cancer treatment. However, the combinations must be based on an understanding of both tumor biology and immune biology.
The involvement of scientists with primary training in physical, engineering and chemical disciplines brings exceptional new capability to the goal of controlling the immune response with nanoparticle-based ISV. The expenses of both preclinical and clinical research mean that industrial resources are mandatory before new strategies can become clinically established, but fortunately industry recognizes the promise and investments are rapidly expanding. While cancer therapy rarely moves into clinical usage rapidly, easily or cheaply, in situ vaccination will likely become a common approach and nanoparticles of various type swill be an important reagent category for ISV.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Am J Med Sci. 1893;10:487–511.
- Coley WB. A report of recent cases of inoperable sarcoma successfully treated with mixed toxins of erysipelas and Bacillus prodigiousus. Surgery, Gynecol Obstet. 1911;13:174.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–2526.
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86.
- Jessy T. Immunity over inability: the spontaneous regression of cancer. J Nat Sci Biol Med. 2011;2(1):43–49.
- Chang S, Kohrt H, Maecker HT. Monitoring the immune competence of cancer patients to predict outcome. Cancer Immunol Immunother. 2014;63(7):713–719.
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
- Mushegian A, Medzhitov R. Evolutionary perspective on innate immune recognition. J Cell Biol. 2001;155(5):705–710.
- Greenwood B. The contribution of vaccination to global health: past, present and future. Philos Trans R Soc Lond, B, Biol Sci. 2014;369(1645):20130433
- Doherty M, Buchy P, Standaert B, et al. Vaccine impact: benefits for human health. Vaccine. 2016;34(52):6707–6714.
- Bloom DE, Canning D, Shenoy ES. The effect of vaccination on children’s physical and cognitivedevelopment in the Philippines. Appl Econ. 2011;44(21):2777–2783.
- Arias-Pulido H, Cimino-Mathews A, Chaher N, et al. The combined presence of CD20 + B cells and PD-L1 + tumor-infiltrating lymphocytes in inflammatory breast cancer is prognostic of improved patient outcome. Breast Cancer Res Treat. 2018;171(2):273–282.
- Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. 2017;14(8):662–674.
- Burnet FM. Immunological recognition of self. Science. 1961;133(3449):307–311.
- Houghton AN, Gold JS, Blachere NE. Immunity against cancer: lessons learned from melanoma. Curr Opin Immunol. 2001;13(2):134–140.
- Ilyas S, Yang JC. Landscape of tumor antigens in T cell immunotherapy. J Immunol. 2015;195(11):5117–5122.
- Klein G. Tumor antigens. Annu Rev Microbiol. 1966;20:223–252.
- Klein G, Klein E. Immune surveillance against virus-induced tumors and nonrejectability of spontaneous tumors: contrasting consequences of host versus tumor evolution. Proc Natl Acad Sci USA. 1977;74(5):2121–2125.
- Lamon EW. The immune response to virally determined tumor associated antigens. Biochim Biophys Acta. 1974;355(2):149–176.
- Bonaventura P, Shekarian T, Alcazer V, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol. 2019;10:168.
- Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6(1):157.
- Byrne KT, Turk MJ. New perspectives on the role of vitiligo in immune responses to melanoma. Oncotarget. 2011;2(9):684–694.
- Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12(1):93.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18(1):128.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421.
- Neefjes J, Jongsma M, Paul P, et al. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836.
- Ward JP, Gubin MM, Schreiber RD. The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. Adv Immunol. 2016;130:25–74.
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473.
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to pd-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128.
- Sharabi A, Kim SS, Kato S, et al. Exceptional response to nivolumab and stereotactic body radiation therapy (sbrt) in neuroendocrine cervical carcinoma with high tumor mutational burden: management considerations from the center for personalized cancer therapy at uc san diego moores cancer center. Oncologist. 2017;22(6):631–637.
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360.
- Sharma P, Hu-Lieskovan S, Wargo JA, et al. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168(4):707–723.
- Chanmee T, Ontong P, Konno K, et al. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers. 2014;6(3):1670–1690.
- Kim J, Bae JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016;2016:6058147.
- Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72(13):3125–3130.
- Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72(9):2162–2171.
- Janikashvili N, Bonnotte B, Katsanis E, et al. The dendritic cell-regulatory T lymphocyte crosstalk contributes to tumor-induced tolerance. Clin Dev Immunol. 2011;2011:430394.
- Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108(5):1435–1440.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174.
- Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200(2):422–431.
- Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. 2016;94(5):509–522.
- Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550.
- Young A, Quandt Z, Bluestone JA. The balancing act between cancer immunity and autoimmunity in response to immunotherapy. Cancer Immunol Res. 2018;6(12):1445–1452.
- Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–580.
- Li B, Chan HL, Chen P. Immune checkpoint inhibitors: basics and challenges. Curr Med Chem. 2019;26(17):3009–3025.
- Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–598.
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218.
- Ishihara D, Pop L, Takeshima T, et al. Rationale and evidence to combine radiation therapy and immunotherapy for cancer treatment. Cancer Immunol Immunother. 2017;66(3):281–298.
- Min Y, Roche KC, Tian S, et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat Nanotechnol. 2017;12(9):877–882.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
- Bouzid R, Peppelenbosch M, Buschow SI. Opportunities for conventional and in situ cancer vaccine strategies and combination with immunotherapy for gastrointestinal cancers. A Review. Cancers. 2020;12(5):1121.
- Sheen MR, Fiering S. In situ vaccination: harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;11(1):e1524.
- JAMA Network. The failure of the erysipelas toxins. JAMA. 1894;24:919.
- Goto T. Radiation as an in situ auto-vaccination: current perspectives and challenges. Vaccines. 2019;7(3):100.
- Formenti SC, Demaria S. Radiation therapy to convert the tumor into an in situ vaccine. Int J Radiat Oncol Biol Phys. 2012;84(4):879–880.
- Hoopes PJ, Moodie KL, Petryk AA, et al. 2017. Hypo-fractionated radiation, magnetic nanoparticle hyperthermia and a viral immunotherapy treatment of spontaneous canine cancer. Paper presented at the Proceedings of SPIE–the International Society for Optical Engineering, 2017 February 22, San Fransisco, CA.
- Hoffman HA, Chakrabarti L, Dumont MF, et al. Prussian blue nanoparticles for laser-induced photothermal therapy of tumors. RSC Adv. 2014;4(56):29729–29734.
- Burga RA, Patel S, Bollard CM, et al. Conjugating Prussian blue nanoparticles onto antigen-specific T cells as a combined nanoimmunotherapy. Nanomedicine. 2016;11(14):1759–1767.
- Cai Z, Xin F, Wei Z, et al. Photodynamic therapy combined with antihypoxic signaling and CpG adjuvant as an in situ tumor vaccine based on metal-organic framework nanoparticles to boost cancer immunotherapy. Adv Healthc Mater. 2020;9(1):e1900996.
- Toraya-Brown S, Fiering S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int J Hyperthermia. 2014;30(8):531–539.
- van den Bijgaart RJ, Eikelenboom DC, Hoogenboom M, et al. Thermal and mechanical high-intensity focused ultrasound: perspectives on tumor ablation, immune effects and combination strategies. Cancer Immunol Immunother. 2017;66(2):247–258.
- Mole R H. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26(305):234–241.
- Nobler MP. The abscopal effect in malignant lymphoma and its relationship to lymphocyte circulation. Radiology. 1969;93(2):410–412.
- Lome LG, Navani S, Aral IM. Spontaneous regression of pulmonary metastases from transitional cell carcinoma of the bladder. Cancer. 1970;26(2):415–418.
- Kingsley DP. An interesting case of possible abscopal effect in malignant melanoma. Br J Radiol. 1975;48(574):863–866.
- Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–870.
- Brix N, Tiefenthaller A, Anders H, et al. Abscopal, immunological effects of radiotherapy: narrowing the gap between clinical and preclinical experiences. Immunol Rev. 2017;280(1):249–279.
- Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111.
- Kroemer G, Galluzzi L, Kepp O, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
- Tesniere A, Panaretakis T, Kepp O, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15(1):3–12.
- Lin FC, Hsu CH, Lin YY. Nano-therapeutic cancer immunotherapy using hyperthermia-induced heat shock proteins: insights from mathematical modeling. Int J Nanomedicine. 2018;13:3529–3539.
- Jego G, Hazoumé A, Seigneuric R, et al. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332(2):275–285.
- Zhou J, Wang G, Chen Y, et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J Cell Mol Med. 2019;23(8):4854–4865.
- Wennerberg E, Lhuillier C, Vanpouille-Box C, et al. Barriers to radiation-induced in situ tumor vaccination. Front Immunol. 2017;8:229.
- Sethuraman SN, Singh MP, Patil G, et al. Novel calreticulin-nanoparticle in combination with focused ultrasound induces immunogenic cell death in melanoma to enhance antitumor immunity. Theranostics. 2020;10(8):3397–3412.
- Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10(426):eaan4488.
- Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696.
- Hammerich L, Marron TU, Upadhyay R, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25(5):814–824.
- Hugander A, Hafström L, Jönsson PE, et al. Local hyperthermia in treatment of experimental liver tumors. Cancer. 1984;53(8):1686–1691.
- Ostapenko VV, Tanaka H, Miyano M, et al. Immune-related effects of local hyperthermia in patients with primary liver cancer. Hepato-gastroenterology. 2005;52(65):1502–1506.
- McDermott S, Gervais DA. Radiofrequency ablation of liver tumors. Semin Intervent Radiol. 2013;30(1):49–55.
- Toraya-Brown S, Sheen MR, Zhang P, et al. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine. 2014;10(6):1273–1285.
- Chen H, Kuo MT. Improving radiotherapy in cancer treatment: promises and challenges. Oncotarget. 2017;8(37):62742–62758.
- Demicheli R, Retsky MW, Hrushesky WJ, et al. The effects of surgery on tumor growth: a century of investigations. Ann Oncol. 2008;19(11):1821–1828.
- Richman SP, Gutterman JU, Hersh EM. Cancer immunotherapy. Can Med Assoc J. 1979;120(3):322–324.
- Old LJ, Clarke DA, Benacerraf B. Effect of Bacillus Calmette-Guerin infection on transplanted tumours in the mouse. Nature. 1959;184(5):291–292.
- deKernion JB, Golub SH, Gupta RK, et al. Successful transurethral intralesional BCG therapy of a bladder melanoma. Cancer. 1975;36(5):1662–1667.
- Lamm DL, Thor DE, Harris SC, et al. Bacillus Calmette-Guerin immunotherapy of superficial bladder cancer. J Urol. 1980;124(1):38–40.
- Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. J Urol. 1976;116(2):180–183.
- Herr HW, Morales A. History of Bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol. 2008;179(1):53–56.
- Rehman H, Silk AW, Kane MP, et al. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer. 2016;4:53.
- Hawkins LK, Lemoine NR, Kirn D. Oncolytic biotherapy: a novel therapeutic platform. The Lancet Oncol. 2002;3(1):17–26.
- Thompson JF, Agarwala SS, Smithers BM, et al. Phase 2 study of intralesional PV-10 in refractory metastatic melanoma. Ann Surg Oncol. 2015;22(7):2135–2142.
- Swift L, Zhang C, Trippett T, et al. Potent in vitro and xenograft antitumor activity of a novel agent, PV-10, against relapsed and refractory neuroblastoma. Onco Targets Ther. 2019;12:1293–1307.
- Liu H, Innamarato PP, Kodumudi K, et al. Intralesional rose bengal in melanoma elicits tumor immunity via activation of dendritic cells by the release of high mobility group box 1. Oncotarget. 2016;7(25):37893–37905.
- Liu H, Weber A, Morse J, et al. T cell mediated immunity after combination therapy with intralesional PV-10 and blockade of the PD-1/PD-L1 pathway in a murine melanoma model. PloS One. 2018;13(4):e0196033.
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12):6387–6392.
- Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79.
- Iyer AK, Khaled G, Fang J, et al. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818.
- Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207.
- Gordon AN, Fleagle JT, Guthrie D, et al. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol. 2001;19(14):3312–3322.
- Nichols JW, Bae YH. EPR: evidence and fallacy. J Control Release. 2014;190:451–464.
- Danhier F. To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244(A):108–121.
- Rawat M, Singh D, Saraf S, et al. Nanocarriers: promising vehicle for bioactive drugs. Biol Pharm Bull. 2006;29(9):1790–1798.
- Holback H, Yeo Y. Intratumoral drug delivery with nanoparticulate carriers. Pharm Res. 2011;28(8):1819–1830.
- Sau S, Agarwalla P, Mukherjee S, et al. Cancer cell-selective promoter recognition accompanies antitumor effect by glucocorticoid receptor-targeted gold nanoparticle. Nanoscale. 2014;6(12):6745–6754.
- Conniot J, Silva JM, Fernandes JG, et al. Cancer immunotherapy: nanodelivery approaches for immune cell targeting and tracking. Front Chem. 2014;2:105.
- Aghebati-Maleki A, Dolati S, Ahmadi M, et al. Nanoparticles and cancer therapy: perspectives for application of nanoparticles in the treatment of cancers. J Cell Physiol. 2020;235(3):1962–1972.
- Sau S, Alsaab HO, Bhise K, et al. Multifunctional nanoparticles for cancer immunotherapy: a groundbreaking approach for reprogramming malfunctioned tumor environment. J Control Release. 2018;274:24–34.
- Lee IH, Kwon HK, An S, et al. Imageable antigen-presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo. Angew Chem Int Ed Engl. 2012;51(35):8800–8805.
- Toraya-Brown S, Sheen MR, Baird JR, et al. Phagocytes mediate targeting of iron oxide nanoparticles to tumors for cancer therapy. Integr Biol. 2013;5(1):159–171.
- Mukherjee S, Ray S, Thakur R. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci. 2009;71(4):349–358.
- Lizotte PH, Wen AM, Sheen MR, et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol. 2016;11(3):295–303.
- Liu D, Poon C, Lu K, et al. Self-assembled nanoscale coordination polymers with trigger release properties for effective anticancer therapy. Nat Commun. 2014;5:4182.
- Vesely MD, Kershaw MH, Schreiber RD, et al. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271.
- Alexis F, Pridgen E, Molnar LK, et al. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515.
- Bertrand N, Wu J, Xu X, et al. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25.
- Fay F, Scott CJ. Antibody-targeted nanoparticles for cancer therapy. Immunotherapy. 2011;3(3):381–394.
- Carter T, Mulholland P, Chester K. Antibody-targeted nanoparticles for cancer treatment. Immunotherapy. 2016;8(8):941–958.
- Rosenblum D, Joshi N, Tao W, et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410.
- Gustafson HH, Holt-Casper D, Grainger DW, et al. Nanoparticle uptake: the phagocyte problem. Nano Today. 2015;10(4):487–510.
- Janßen HC, Angrisani N, Kalies S, et al. Biodistribution, biocompatibility and targeted accumulation of magnetic nanoporous silica nanoparticles as drug carrier in orthopedics. J Nanobiotechnology. 2020;18(1):14.
- Lança T, Silva-Santos B. The split nature of tumor-infiltrating leukocytes: implications for cancer surveillance and immunotherapy. Oncoimmunology. 2012;1(5):717–725.
- Mittal D, Gubin MM, Schreiber RD, et al. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25.
- Sheen MR, Lizotte PH, Toraya-Brown S, et al. Stimulating antitumor immunity with nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014;6(5):496–505.
- Awate S, Babiuk LA, Mutwiri G. Mechanisms of action of adjuvants. Front Immunol. 2013;4:114.
- Lee KL, Murray AA, Le DHT, et al. Combination of plant virus nanoparticle-based in situ vaccination with chemotherapy potentiates antitumor response. Nano Lett. 2017;17(7):4019–4028.
- Lee JM, Lee MH, Garon E, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8+ T-cell infiltration. Clin Cancer Res. 2017;23(16):4556–4568.
- Le DH, Lee KL, Shukla S, et al. Potato virus X, a filamentous plant viral nanoparticle for doxorubicin delivery in cancer therapy. Nanoscale. 2017;9(6):2348–2357.
- Amarante-Mendes GP, Adjemian S, Branco LM, et al. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol. 2018;9:2379.
- Rosenfeld MR, Chamberlain MC, Grossman SA, et al. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro-Oncology. 2010;12(10):1071–1077.
- Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. 2015;9(10):1966–1981.
- Salazar AM, Erlich RB, Mark A, et al. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, and clinical trial. Cancer Immunol Res. 2014;2(8):720–724.
- Gopalakrishnan V, Helmink BA, Spencer CN, et al. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–580.
- Fessler J, Matson V, Gajewski TF. Exploring the emerging role of the microbiome in cancer immunotherapy. J Immunother Cancer. 2019;7(1):108.
- Roger A, Finet A, Boru B, et al. Efficacy of combined hypo-fractionated radiotherapy and anti-PD-1 monotherapy in difficult-to-treat advanced melanoma patients. Oncoimmunology. 2018;7(7):e1442166.
- Hammerich L, Davis TA, Keler T, et al. Combining in situ vaccination with immune checkpoint blockade induces long-term regression of lymphoma tumors. Blood. 2016;128(22):465–465.
- Khalil DN, Suek N, Campesato LF, et al. In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J Clin Invest. 2019;129(8):3435–3447.
- Atkins MB, Robertson MJ, Gordon M, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997;3(3):409–417.
- Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–2548.
- Fiszer-Maliszewska L, Den Otter W, Madej JA, et al. Therapeutic potential of biological response modifiers against transplantable mouse tumors of spontaneous origin. II. Local interleukin 2 treatment of tumors of different immunogenic strength. Arch Immunol Ther Exp. 1998;46(5):293–300.
- Radny P, Caroli UM, Bauer J, et al. Phase II trial of intralesional therapy with interleukin-2 in soft-tissue melanoma metastases. Br J Cancer. 2003;89(9):1620–1626.
- Weide B, Derhovanessian E, Pflugfelder A, et al. High response rate after intratumoral treatment with interleukin-2: results from a phase 2 study in 51 patients with metastasized melanoma. Cancer. 2010;116(17):4139–4146.
- Spitzer MH, Carmi Y, Reticker-Flynn NE, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168(3):487–502.