
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Sub-ablative heat induces pleiotropic biological effects in cancer cells, activating programmed cell death or survival processes. These processes decide the fate of the heated cell. This study investigates these and assesses whether heat, in combination with HSP90 inhibition, augments cell death and induces a pro-immune phenotype in these cells.
Methods
HCT116 and HT29 cells were subjected to thermal doses (TID) of 60 and 120CEM43 using a PCR thermal cycler. HSP90 was inhibited with NVP-AUY922. Viability was assessed using the MTT assay. Cellular ATP and HSP70 release were assessed using ATP and Enzyme-linked Immunosorbent assays, respectively. Flow cytometry and immunoblotting were used to study the regulation of biomarkers associated with the heat shock response, the cell cycle, and immunogenic and programmed cell death.
Results
Exposure of HCT116 and HT29 cells to TIDs of 60 and 120CEM43 decreased their viability. In addition, treatment with 120CEM43 increased intracellular HSP70 and the percentage of HCT116/HT29 cells in the G2/M cell cycle phase, ATP release and Calreticulin/HSP70/HSP90 exposure in the plasma membrane, while downregulating CD47 compared to sham-exposed cells. When combined with NVP-AUY922, treatment of HCT116/HT29 cells with 120CEM43 resulted in a synergistic decrease of cell viability associated with the induction of apoptosis. Also, the combined treatments increased Calreticulin exposure, CD47 downregulation, and HSP70 release compared to the sham-exposed cells.
Conclusion
Sub-ablative heating can act synergistically with the clinically relevant HSP90 inhibitor NVP-AUY922 to induce a pro-immunogenic form of cell death in colon cancer cells.
Introduction
Colon cancer is one of the most common cancers worldwide, with approximately 1.9 million new cases being diagnosed every year [Citation1]. Treatment options depend on the location and staging of the tumors, and typically involve combinations of surgery, radiotherapy, and chemotherapy. Despite the improved mortality rates, over 935,000 people died worldwide in 2020 from this disease [Citation1], and hence effective novel therapies are urgently needed.
Biophysical thermal treatment modalities such as high intensity focused ultrasound (HIFU) [Citation2], radiofrequency [Citation3], microwave [Citation3], and laser ablation [Citation4,Citation5] have attracted clinical interest due to their noninvasive (or minimally-invasive) and cost-effective nature [Citation6]. They raise the temperature in the target tissue and have shown clinical therapeutic benefit in neurosurgery [Citation7], ophthalmology [Citation8], gynecology [Citation9], and oncology where they have been used for the treatment of benign and malignant tumors of the uterus [Citation10], prostate [Citation11,Citation12], breast [Citation13], liver [Citation14] and pancreas [Citation15]. Heat can kill tumor cells by inducing necrosis, activating programmed cell death, and provoking an immune response [Citation16–19]. Immunogenic cell death (ICD) is a form of apoptosis that is associated with the activation of the immune system in vivo [Citation20,Citation21]. Calreticulin (CALR) exposure on the plasma membrane [Citation22], ATP release [Citation23], and heat shock protein (including HSP70 and HSP90) translocation to the plasma membrane [Citation24], and CD47 downregulation are amongst its hallmarks [Citation25,Citation26].
Thermal exposures delivered at different temperatures for varying lengths of time can be described using a parameter known as the Thermal Isoeffective Dose (TID) for which the units are in terms of cumulative equivalent minutes at 43 °C (CEM43). It is described mathematically by:
(1)
(1)
where TID (CEM43) is the thermal dose in units of equivalent minutes at 43 °C (CEM43), dt is the time at temperature T (oC), RCEM is a constant approximated to 0.25 for temperatures below, and 0.5 for temperatures above 43 °C.
represents the final time point of the thermal treatment [Citation27]. A TID of 240 CEM43 is taken to be the threshold for effective cell killing in vivo [Citation28] but preclinical evidence has shown that not all heated cells may die at this level [Citation29]. Thermotolerance, defined as acquired resistance to heat, develops in these cells and is mediated by the activation of the heat stress response to counter cell death mechanisms [Citation17], including that of heat-induced protein denaturation [Citation30–32]. These pro-survival processes are thought to be mediated by heat shock proteins such as HSP70 and HSP90 which engulf misfolded and damaged proteins to facilitate their appropriate folding [Citation33,Citation34] or to target them for degradation [Citation35,Citation36].
HSP70 and HSP90 belong to the heat shock protein family discovered in 1962 [Citation37]. They are ubiquitously expressed and are named after their molecular weights. HSP70 is expressed in the cytosol and nucleus, it is temperature-sensitive, and its expression is increased in response to heat stimuli. HSP90 is one of the most abundant proteins in cells, comprising ∼1% of the total protein content [Citation38]. It is necessary for cell survival, and maintains cellular protein homeostasis, acting as a molecular chaperone to help folding and trafficking of over 400 protein clients. Conformational changes driven by ATP hydrolysis, with an ATP binding domain located in its N-terminal domain, are essential for carrying out its functions [Citation39–41]. The already ubiquitous expression of HSP90 in normal cells is higher in cancer cells, including those of colon origin [Citation42]. Differential affinity binding between HSP90 and its protein clients in normal and cancer cells, possibly due to post-translational modifications affecting HSP90, and differential localization of HSP90 with more protein located in the plasma membrane of cancer cells relative to that of normal cells, have been postulated for the significance of HSP90 in cancer cell survival [Citation43]. The immune-modulatory effects of HSP90 have attracted increased attention with its inhibition increasing the susceptibility of tumors to immune checkpoint immunotherapies [Citation44], and also reducing graft versus host disease, in part, by contributing to T cell apoptosis and the reduction of cytokine release [Citation45]. For these reasons, HSP90 has been suggested as a potential therapeutic target in cancer.
HSP90 inhibitors first entered phase I clinical trial in 1999 [Citation46–48]. The limitations of initial compounds included poor solubility, limited bioavailability, and hepatotoxicity [Citation49–51]. NVP-AUY922 (AUY922) is an HSP90 inhibitor developed using the 4,5-diarylisoxazole scaffold, which has shown high cellular uptake and potency against HSP90 by binding to the NH2-terminal domain of the protein [Citation52,Citation53]. AUY922 has shown clinical benefit in a phase 2 trial of patients with refractory gastrointestinal stromal tumors. Patients receiving AUY922 had progression-free survival of 4 months. This compared favorably with historical placebo controls for whom this was 6 weeks [Citation54]. A phase 1/1 b trial of AUY922 as monotherapy in patients with relapsed or refractory multiple myeloma resulted in 16 out of 24 patients showing stable disease [Citation55]. In another phase II clinical trial of 29 patients with non-small cell lung cancer characterized by EGFR exon20 insertions, AUY922 was well tolerated and patients had a median progression-free survival of 2.9 months and median overall survival of 13 months. This exceeded the pre-determined target efficacy [Citation56].
Pre-clinical efforts to enhance the anti-cancer effects of HSP90 inhibitors by combining them with heat are ongoing. For example, ‘mild hyperthermic’ exposure of melanoma tumors at 43 °C for 30 min (TID of 30CEM43) combined with both 17-DMAG and quercetin, inhibitors of HSP90 and HSP70 respectively, but not with 17-DMAG alone improved tumor growth control compared to heated only subjects [Citation57]. Clonogenic survival of SiHa cervix cancer cells, compared to that of cells treated with Ganetespib alone, was also decreased in a statistically significant manner when 30 nM of this HSP90 inhibitor was used in combination with a TID of 15CEM43. These authors used elegant experiments on SiHa and HeLa cells to show the molecular effect of ‘mild hyperthermic’ heating (42 °C for 60 min (15 CEM43)) on DNA damage as well as their chemo and radio-sensitization [Citation58]. Induction of ICD within 24 h of exposure to an ablative TID of 1,000CEM43 (47 °C/1 h), but not to a TID of 15CEM43 (42 °C for 60 min) [Citation59] has been shown in lung carcinoma A549 and metastatic ovarian OV90 cell lines in the absence of heat shock protein inhibition. Thus it remains unclear whether sub-ablative heat (e.g., TIDs of 60 and 120 CEM43) in the presence or absence of an HSP90-specific inhibitor such as AUY922 can induce sustained pro-immune and synergistic cytotoxic effects in cancer cells. This is the subject of the current study where the response of the HCT116 and HT29 colon cancer cells to these treatments is characterized longitudinally by examining biomarkers associated with survival (intracellular heat shock protein expression, cell cycle regulation) and ICD (ATP release, exposure of CALR, HSP70, HSP90 and CD47 on the plasma membrane). Data are presented, to show not only the dynamic response of the cancer cells to these treatments and how HSP90 inhibition and sub-ablative heat synergize to augment their anti-cancer effects but also to contribute toward a paradigm shift in thermal research where specific biological mechanisms are induced by a ‘narrow’ range of TIDs.
Materials and methods
Cell culture
HCT116 and HT29 cells were purchased from LGC standards (Teddington, UK) and maintained as sub-confluent monolayers at 37 °C in a humidified atmosphere containing 5% CO2. They were propagated using McCoys5A supplemented with 10% FBS, 2 mM L-glutamine, and sub-cultured using Accutase (SIGMA, UK). Screening for mycoplasma contamination was carried out monthly. For experiments, cells of low passage (<20) from culture were detached and 60 µl, at a cell density of 106 cells/ml were transferred to PCR tubes at 4 °C for thermal treatment.
Thermal exposures and thermometry
Cells in suspension were placed in 0.2 ml polymerase chain reaction (PCR) tubes (Corning Inc., USA), and heated using a PCR thermal cycler (Biorad, Hercules, USA) ( and Supplement (a)). In this system, the temperature of the thermal block could be pre-programmed (thermal accuracy: ±0.3 °C at 90 °C; thermal uniformity: ±0.4 °C well-to-well at 90 °C; ramping speed: 3 °C/sec; power: 850 W; 0.1 °C step size). For all exposures the TID delivered was within 10% of that intended as described previously [Citation29,Citation60].
Table 1. Thermal exposures were used in this study.
Cell viability: MTT assay
An MTT assay was used to determine (a) the viability of treated cells relative to that of the sham-exposed cells, or (b) the number of live cells by extending this assay to account for the MTT standard curves of cell number versus optical density. The MTT assay is based on the color change seen when the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide is reduced to formazan in mitochondria. The intensity of the signal from the assay correlates with live cell number over a range of at least 0–2 × 106 cells/ml. To estimate the relative viability of cells at the end of their treatments, 20 µl MTT dye (5 mg/ml)/100 µl of the medium was pipetted into each well of a 96-well plate containing the sham-exposed or treated cells. The plate was incubated at 37 °C for 2 h. 100 µl of 10% sodium dodecyl sulfate (SDS) was then added to each well. After overnight incubation at 37 °C, the signal at 550 nm (and a reference signal at 630 nm) was detected using a colorimetric microplate reader. Then, the background signal was subtracted from the optical density measurements. Results for treated cells were normalized to those of sham-exposed cells and were expressed as a percentage to provide a measure of the relative cell viability for each condition. To calculate the number of viable cells (e.g., ) MTT standard curves of cell number versus optical density were obtained in addition to the MTT assay described above. These MTT standard curves were obtained by plating increasing densities (1.2 × 103 cells/well to 2 × 105 cells/well counted using a hemocytometer, 4 replicates/condition) of HCT116 and HT29 cells in the wells of a 96-well plate. The cells were allowed to settle for 8 h and were then assayed using MTT alongside those undergoing experimental conditions. In addition, background optical density signal from wells containing medium and MTT reagents, but no cells, was measured and subtracted from each condition. MTT standard curve data were visualized in Excel which enabled the plotting of linear trendlines for both cell lines (0 to 2 × 105 cells), the automatic calculation of their R2 values, and cell number versus optical density equations. An example of these standard curves is shown in Supplement Figure S1. Thus, by rearranging the equations for the number of viable cells (the ‘x’ component of these equations) their number was calculated for every MTT optical density measurement in the experimental conditions.
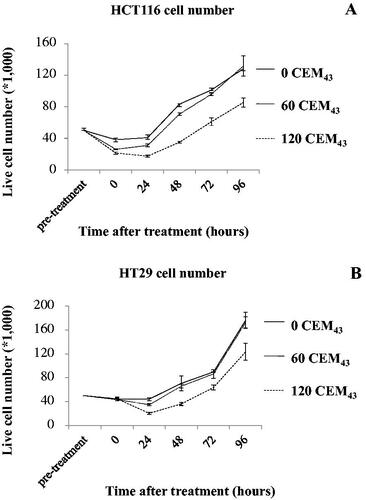
Figure 1. The number of live HCT116 (A) and HT29 (B) cells immediately before, and after, treatment with TIDs of 0, 60, and 120CEM43 up to 96 h. Results are presented as average ± Std. dev of 4 replicates within experiments that have been performed 3 times with similar results.
Combination index (CI) calculation
To determine whether the effects of the combined heat and AUY922 treatments in these colon cancer cells were antagonistic, additive, or synergistic, a CI-based method (Chou-Talalay) was used [Citation61]. Here the treatment dose to achieve a given reduction in cell viability when used in combination was compared to those required to achieve the same viability level if each treatment was given alone. This was done by calculating the CI for these treatments. A CI of < 1, > 1, = 1 denotes synergism, antagonism, or additivity between 2 treatments, respectively. In its simplified form, the CI can be given mathematically by:
(2)
(2)
where TD1X represents the dose of the 1st treatment that results in a reduction of cell viability of x% if used alone, TD2X represents the dose of the 2nd treatment giving the same reduction of cell viability of x% if used alone, and TD1 and TD2 are the doses of the 2 treatments required to provide the same effect when used in combination.
ATP release in the supernatant
ATP in the supernatant of control and thermally treated cells was measured using an ATP detection kit (Thermofisher Scientific ltd., Waltham, USA). 100 µl of supernatant was collected and centrifuged at 200 g to spin down cell debris. The supernatant was then used to determine the amount of ATP present according to the manufacturer’s instructions. 10 mM D-luciferin solution was prepared using stocks provided in the kit and was protected from light until use. 100 mM DTT solution was prepared by adding 1.62 ml of distilled water to 25 mg of DTT. The reaction mixture was prepared by combining 8.9 ml distilled water, 0.5 ml of the reaction buffer provided in the kit, 0.1 ml 0.1 M DTT, 0.5 ml 10 mM D-luciferin, and 2.5 µL of firefly luciferase (5 mg/mL stock solution). The tube was gently inverted to mix. This reaction solution was mixed with the test supernatant with the amount of sample added amounting to no more than 10% of the total assay volume. The luminescence was then read using a luminescent microplate reader.
HSP70 and HMGB1 release in the supernatant
The colorimetric HSP70 High Sensitivity Kit (cat. number: ab133061, Abcam (Cambridge, UK)) and the HMGB1 enzyme-linked immunosorbent assay (ELISA) kit (cat. number: NBP2-62766, Novus Biologicals (Colorado, USA)) were used to investigate the levels of HSP70 and HMGB1 release in the supernatant of cells, following the manufacturers’ instructions. Briefly, supernatants were collected from sham-exposed and treated cells and were centrifuged at 200 × g. Tests were carried out in duplicates by pipetting 100 µl of the supernatant for each condition into 96-well plates pre-coated with the test antibody. Wells were then washed, incubated with the capture and detection antibodies, and washed again with the buffers provided in the kits, as described in the product manuals. For a signal generation, the wells were incubated with the TMB reaction solutions, and color was measured using a colorimetric plate reader at 450 nm.
Regulation of protein and cell cycle analysis: immunoblotting and flow cytometry
Immunoblotting was used to determine the intracellular levels of HSP70, HSP90, cleaved PARP, cleaved caspase 3, cleaved caspase 9, and LC3A () as described in Supplement (b). Semi-quantitative analysis of the immunoblotting signal was performed using the Image J image analysis software to provide densitometry data for each blot. For flow cytometry analysis of protein expression on the plasma membrane, cell cycle analysis and induction of apoptosis using Annexin V/Propidium Iodide (PI) staining flasks were washed with PBS, cells were detached from flasks using Accutase, washed with PBS, and stained as described in Supplement (c). The cells expressing ‘high’ levels of the proteins were arbitrarily defined by determining the fluorescence intensity threshold that would result in ∼5% of cells exposed to a TID of 0CEM43 being gated as high fluorescence populations in the corresponding histogram plots. These fluorescence intensity thresholds were then applied to the histogram plots of the cells treated with TIDs of 60 and 120CEM43. The percentage of cells positive for fluorescent intensities exceeding these thresholds was then calculated. The cells expressing ‘low’ levels of CD47 protein were defined as shown in Supplement Figure S2. Apoptotic cells were defined as Annexin V positive and PI negative, and are shown in the bottom right quadrangle of the Annexin V FITC/PI plots.
Figure 2. The regulation of biomarkers associated with ICD in HCT116 and HT29 cells exposed to TIDs of 0, 60, and 120CEM43 24 and 48 h after treatment. (A,B) ATP release from HCT116 and HT29 cells. (C,D) Percentage of HCT116 and HT29 cells positive for CALR exposure on their plasma membrane. (E) Percentage of HCT116, and (F) percentage of HT29 cells positive for HSP70 protein on their plasma membrane. (G) Percentage of HCT116, and (H) percentage of HT29 cells positive for HSP90 protein on their plasma membrane. (I-J) Percentage of HCT116 and HT29 cells expressing ‘low’ levels of CD47 on their plasma membrane. Results are presented as means ± Std. dev of at least n = 3 independent experiments. Statistical significance where it exists between cells exposed to a TID of 60 and/or 120CEM43, and cells exposed to a TID of 0CEM43 is denoted with an asterisk ‘, and is assumed at p < .05.
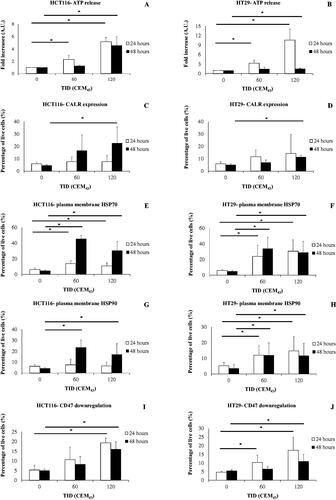
Table 2. Antibodies were used in this study.
Statistical analysis of results
Results for the cell number assay () are presented as means ± standard deviation (Std. dev) of 4 data sets of 1 experiment that has been replicated 3 times with similar results. Results for all other experiments (ATP, HSP70, and HMGB1 release assays, immunoblotting, flow cytometry, and all other viability data) are presented as means ± Std. dev of at least 3 independent experiments. Statistical significance was assessed using the Student’s T-test, assuming a one-tail distribution two-sample unequal variance. p < .05 was considered to be significant.
Results
Effect of heat on the viability of Colon cancer cells
HCT116 and HT29 cells were treated with TIDs of 0, 60, 120CEM43 () and the number of viable cells were determined immediately using the MTT-based method described above, and then daily up to 96 h after treatment. Results showed that out of the 50 × 103 HCT116 cells (counted using a hemocytometer) plated before treatment 38 ± 3 × 103 cells sham-exposed to heat remained alive immediately after treatment. These then grew to 41 ± 2 × 103, and 130 ± 6 × 103 24 and 96 h after treatment, respectively (). Treatment of these cells with a TID of 60CEM43 decreased the number of cells to 26 ± 0.5 × 103 and 31 ± 2 × 103 cells immediately, and 24 h after, treatment, respectively. Cell number then increased up to 130 ± 12 × 103 96 h after treatment. Exposure to a TID of 120CEM43 caused a decline in the number of HCT116 cells to 21 ± 1 × 103 immediately after treatment, with a further decline to 17 ± 1.5 × 103 cells 24 h later. Their number then increased up to 85 ± 6 × 103 96 h after treatment (). Similar results were seen for thermally treated HT29 cells (). These results show the dynamic regulation of the survival of colon cancer cells following ‘sub-ablative’ heat, with an initial reduction in their number followed by proliferation of the survivors who overcome the cytotoxic effects.
Effect of heat on the regulation of biomarkers associated with ICD
In order to identify biological mechanisms associated with this response, biomarkers associated with ICD (ATP release, CALR, HSP70, and HSP90 plasma membrane exposure, and CD47 downregulation) were investigated. ATP release in the supernatant of HCT116 and HT29 cells exposed to TIDs of 60 and 120CEM43 was increased 24 h after treatment relative to controls (TID = 0CEM43). When HCT116 and HT29 cells were treated with a TID of 120CEM43 their ATP release was statistically significantly increased 4.5 ± 1.4 and 1.5 ± 0.2 fold respectively 48 h after treatment relative to sham-exposed cells (). Also, 48 h after treatment statistically significant increases in CALR exposure of heated cells was seen, with CALR being detected on the plasma membrane of 23 ± 13% of 120CEM43-exposed HCT116 (), and 11 ± 2% of 120CEM43-exposed HT29 cells (), but only in 5 ± 1% of sham-exposed cells.
A statistically significant enhancement in the plasma membrane localization of HSP70 and HSP90 in HCT116 and HT29 cells was detected after heat treatment. Plasma membrane HSP70 was detected in 45 ± 5% and 30 ± 12% of HCT116 cells (), and 34 ± 14% and 29 ± 14% of HT29 cells () exposed to TIDs of 60 and 120CEM43 respectively, 48 h after treatment. At the same time point plasma membrane HSP90 was detected in 23 ± 7%, 17 ± 10% of HCT116 cells () and 12 ± 8%, 12 ± 8% of HT29 cells () exposed to 60 and 120CEM43 respectively. In contrast, no more than 5 ± 1% of sham-exposed cells (TID = 0CEM43) showed HSP70 and HSP90 on their plasma membrane.
Increases in the percentages of HCT116 and HT29 cells with down-regulated CD47 levels on their plasma membrane were seen after heat exposure. For example ‘low’ (down-regulated) CD47 levels were detected in 16 ± 4% of HCT116 and 11 ± 4% of HT29 cells 48 h after their exposure to TID of 120CEM43, whereas no more than 6 ± 1% of sham-exposed cells had a similar CD47 regulation pattern (). These results show an induction of biomarkers associated with ICD in colon cancer cells exposed to sub-ablative heat.
Effect of heat on the regulation of biomarkers associated with survival
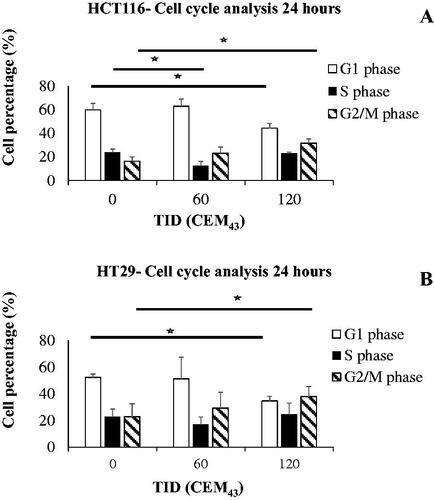
To investigate how cells adapt to the thermal stress caused by these heat exposures the intracellular expression of HSP70 and HSP90, and the distribution of the cells in different phases of the cell cycle were investigated. Increased immunoblotting signal for intracellular HSP70 in HCT116 and HT29 cells was seen 24 h after their treatment with heat (). The densitometry of these blots showed statistically significant increases in HSP70 of 2.5 ± 1.1 and 2.3 ± 1 fold (HCT116) and 2.3 ± 0.7 fold (HT29) in cells exposed to TIDs of 60 (HCT116 only) and 120 CEM43 respectively over the HSP70 levels seen in sham-exposed cells. In contrast, no statistically significant differences were seen in any of the treatment conditions in intracellular HSP90 in HCT116 and HT29-heated cells (). An increase in the percentages of 120CEM43-exposed cells in the G2/M phase of the cell cycle (32 ± 2% of HCT116 and 38 ± 3% of HT29 cells) were seen compared to those of sham-exposed cells (16 ± 2% of HCT116 and 23 ± 3% HT29 cells) 24 h after treatment (). These results show that molecular responses associated with survival are induced in both HCT116 and HT29 cells treated with TID of 120CEM43. Thus we sought to investigate whether inhibiting the cell’s heat shock mechanism with the HSP90 inhibitor AUY922 would increase cell death.
Figure 3. The effect of heat on the regulation of intracellular HSP70 and HSP90. (A) Representative immune-blot images of HSP70, HSP90, and ACTIN in HCT116 and HT29 cells exposed to 0, 60, and 120CEM43 24 h after treatment. (B,C) densitometry data of immunoblot images are shown for HSP70 and HSP90 in HCT116 cells (B) and HT29 cells (C). Data are presented as averages ± Std. dev of at least n = 3 independent experiments. Statistical significance where it exists between cells exposed to a TID of 60 and/or 120CEM43, and cells exposed to a TID of 0CEM43 is denoted with an asterisk ‘⋆‘, and is assumed at p < .05.
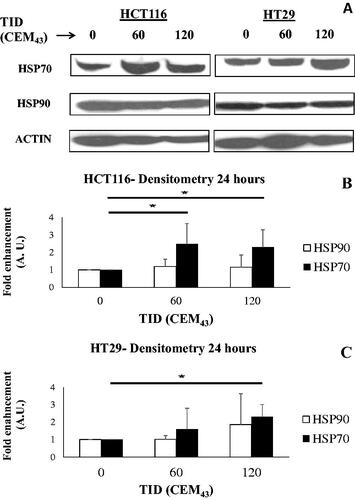
Figure 4. The effect of heat on the regulation of the cell cycle in colon cancer cells. Results of HCT116 (A) and HT29 (B) cell cycle analysis 24 h after treatment with TIDs of 0, 60, or 120CEM43. Results are presented as means ± Std. dev of n = 3 independent experiments. Statistical significance where it exists between cells exposed to a TID of 60 and/or 120CEM43 and sham-exposed cells is denoted with an asterisk ‘⋆‘, and is assumed at p < .05.
The effect of HSP90 inhibition on the viability of heated cells
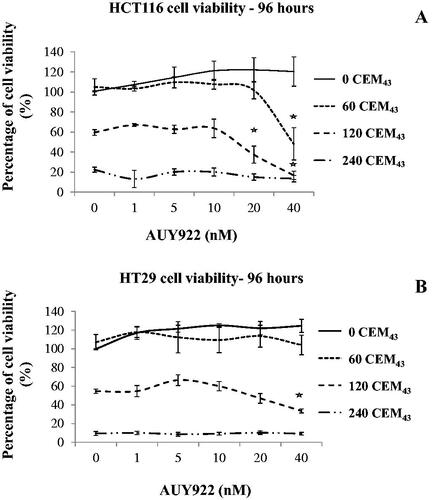
HCT116 and HT29 cells were treated with a combination of heat (TID 0, 60, 120, and 240CEM43) and increasing concentrations of the HSP90 inhibitor AUY922 (0–40nM). A statistically significant decrease in HCT116 and HT29 cell viability was seen after treatment of these cells with a TID of 120CEM43 and 40 nM of AUY922 (16 ± 7% and 33 ± 4% respectively) compared to that of the 120CEM43–only treated HCT116 and HT29 cells (60 ± 5% and 55 ± 5% respectively) (all conditions normalized for the viability of their sham-treated controls (TID = 0CEM43, AUY922 = 0nM), 96 h after treatment (). These results did not depend on the temperature required to achieve a TID of 120CEM43 since the exposure of the cells to 43 °C for 120 min or 46 °C for 15 min (with or without AUY922) resulted in non-statistically significant differences in cell viability (Supplement Figure S3). In addition, a statistically significant decrease in HCT116, but not HT29 cells, was seen when these cells were treated with a TID of 60CEM43 and AUY922 (40 nM) (50 ± 27% and 100 ± 20%, respectively) when compared to the 60CEM43 exposed- HCT116/HT29 cells (100 ± 14% and 105 ± 15%, respectively). Treatment with ablative TIDs (e.g., 240CEM43) resulted in almost complete loss of cell viability (≥80%), and no further decrease in cell viability was seen upon the combination with AUY922 (40 nM) ().
Figure 5. The effect of AUY922 on the viability of heated cells. HCT116 (A) and HT29 cells (B) were exposed to TIDs of 0, 60, 120, and 240CEM43 in the presence of increasing concentrations of AUY922 (0–40 nM). Viability was assessed using the MTT assay 96 h later and results were normalized for the viability of the sham-exposed cells, i.e., those treated with a TID of 0CEM43 in the absence of AUY922. Data are presented as means ± Std. dev of n = 3 independent experiments and statistical significance where it exists for the combined AUY922 and heat treatments compared to the cells exposed to the thermal treatment alone (i.e., no AUY922) is denoted with an asterisk ‘⋆‘, and is assumed at p < .05. For clarity statistical significance has not been shown for cells exposed to heat when compared to the cells sham-exposed to a TID of 0 CEM43.
The CI (EquationEquation 2(2)
(2) ) was used to determine whether the statistically significant reduction in viability in both cell lines exposed to sub-ablative heat (120 CEM43) and AUY922 (40 nM) was due to a synergism between the 2 treatments. AUY922 dose-response curves were obtained, and the dose-response of heated cells (where not already known) was determined (Supplement Figure S4). The CI was calculated to be ≤0.75 for both cell lines (). These results suggest that a ‘sub-ablative’ TID of 120 CEM43 and AUY922 synergize to decrease the viability of these cells.
Table 3. Calculation of the combination index (CI) of the combined heat and AUY922 treatments of HCT116 and HT29 cells.
The effect of HSP90 inhibition on PCD and heat shock protein expression of heated cells
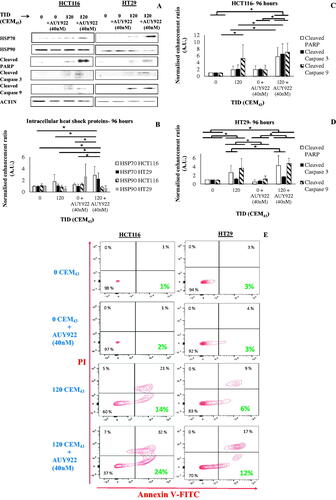
The synergism seen in cells after combined treatments were characterized at the protein level. Statistically significant increases in the intracellular levels of HSP70 (3 ± 1% and 2 ± 1% fold-over sham-exposed cells) were seen in HCT116 and HT29 cells respectively 96 h after their exposure to a TID of 120 CEM43 and AUY922 (40 nM) when compared to all other treatments (sham-exposure, heat-only treatment, and AUY922-only treatment) (). In contrast, no significant changes were seen in intracellular HSP90 regulation in cells treated with heat and/or AUY922 at the same time point (). Then the cytotoxic response was characterized by investigating the intracellular levels of cleaved PARP, cleaved caspase 3, and cleaved caspase 9. HCT116 cells treated with the combined TID of 120 CEM43 and AUY922 (40 nM) showed statistically significant increases in cleaved PARP (6 ± 3% fold-over sham-exposed cells) and cleaved caspase 3 (7 ± 3% fold-over sham-exposed cells) when compared to all other treatments 96 h after treatment (). At the same time point increased levels of cleaved caspase 9 were detected in those cells when treated with the combined heat and AUY922 treatments (7 ± 3% fold-over sham-exposed cells), and when treated with 120 CEM43 only (5 ± 4% fold-over sham-exposed cells) relative to sham-exposed and AUY922-treated cells (). Increases in cleaved PARP, cleaved caspase 3, and cleaved caspase 9 were also seen in HT29 cells treated with a TID of 120 CEM43 and AUY922 (40 nM) compared to sham–exposed, 120 CEM43-treated (cleaved caspase3 and cleaved PARP), and AUY922 (40 nM)-treated cells 96 h after treatment (). Apoptosis induction was confirmed with flow cytometry in both cell lines treated with the combined TID of 120 CEM43 and AUY922 (40 nM). These results showed an increase of Annexin V positive/PI negative HCT116 and HT29 cells treated with 120CEM43 and AUY922 when compared to all other control and treated groups (). Furthermore, non-statistically significant differences were detected in the immunoblotting assessment of LC3A (suggestive of autophagy) in HCT116 and HT29 cells 96 h after their exposure to TIDs of 120CEM43 and AUY922 when compared to all other treatments (Supplement Figure S5). These data show that the reduction in cell viability seen when AUY922 is combined with a TID of 120 CEM43 is, at least in parts, due to cytotoxicity and the induction of PCD in these cells. The effect of the combined treatments on the regulation of pro-immune biomarkers of the cells was then investigated.
Figure 6. The effect of heat and HSP90 inhibition on PCD and heat shock protein regulation. (A) Representative immune-blot images of intracellular HSP70, HSP90, cleaved PARP, cleaved caspase 3, cleaved caspase 9, and ACTIN in HCT116 and HT29 cells exposed to 0 or 120CEM43 in the presence or absence of AUY922 (40 nM) 96 h after treatment. (B-D) densitometry data of the immunoblot images are shown for HSP70 and HSP90 in HCT116 cells and HT29 cells (B), cleaved caspase 3, cleaved caspase 9, and cleaved PARP in HCT116 (C) and HT20 (D) cells. (E) detection of phosphatidylserine exposure on the cell membrane using Annexin V/PI staining 96 h after the exposure of HCT116 and HT29 cells to 0 or 120CEM43 in the presence or absence of AUY922. Apoptotic cells are shown in the bottom right quadrangle to show Annexin V positive/PI negative cells. Data in (B-D) are presented as averages ± Std. dev of at least n = 3 independent experiments and results for cells treated with heat and/or AUY922 have been normalized for that of the control cells (TID = 0CEM43, AUY922 = 0 nM). Statistical significance where it exists is denoted with an asterisk ‘⋆‘, and is assumed at p < .05. Data in (A) and (E) are presented as representative images of experiments that have been repeated at least 3 times (in (E) twice) with similar results.
The effect of HSP90 inhibition on the regulation of pro-immune biomarkers
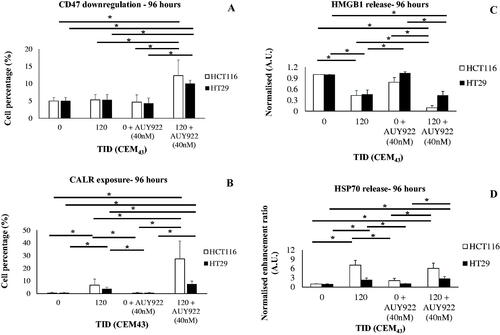
Treatment of HCT116 and HT29 cells with TIDs of 120CEM43 in the presence of AUY922 (40 nM) resulted in a statistically significant increase in the percentage of HCT116 and HT29 cells with downregulated CD47 on their plasma membrane (12 ± 5% and 10 ± 1%, respectively) when compared to sham exposed cells (5 ± 1% and 5 ± 1%, respectively), AUY922 (40 nM)-treated cells (4 ± 2% and 4 ± 1%, respectively) and 120CEM43-treated cells (5 ± 1% and 5 ± 2%, respectively) 96 h after treatment (). In addition, statistically significant increases in CALR exposure were seen in HCT116 and HT29 cells (27 ± 14% and 8 ± 3%, respectively) when compared to the sham-exposed cells (1 ± 1%) and those exposed to a TID of 120 CEM43 (7 ± 5% and 4 ± 1%, respectively) or treated with AUY922 alone (1 ± 1%) 96 h after treatment (). Decreases of 90 ± 5% and 60 ± 10% compared to sham controls were seen in HMGB1 release in the supernatant of HCT116 and HT29 cells respectively 96 h after their treatment with a TID of 120 CEM43 and AUY922 (40 nM). A statistically significant reduction in HMGB1 in HCT116 and HT29 cells were also seen for these treatments when compared to the thermally-exposed (120CEM43) (HCT116 cells only) or AUY922-treated (40 nM) cells (both cell lines). Also, thermal 120CEM43 treatment of HCT116 and HT29 cells decreased the release of HMGB1 in the supernatant by 45 ± 10% compared to shams 96 h after treatment (). Finally, HCT116 and HT29 cells treated with combined 120CEM43 and AUY922 (40 nM) released 6 ± 2% (HCT116) and 3 ± 1% (HT29) fold more HSP70 in their supernatant than sham-exposed cells and AUY922-treated cells. HCT116 and HT29 cells that were only thermally exposed (TID of 120CEM43) released more HSP70 (7 ± 2% and 2 ± 1% fold respectively) than sham-exposed cells (). These results suggest that sub-ablative heat treatment alone not only increased pro-immune signals but that this regulation was stronger when the heat was combined with AUY922.
Figure 7. The effect of heat and HSP90 inhibition on the regulation of immune biomarkers. The percentage of HCT116 cells and HT29 cells treated with 0 and 120CEM43 in the presence or absence of AUY922 (40 nM) with ‘low’ levels of CD47 (A) or CALR expression (B) on their plasma membrane, and the release of HMGB1 (C) and HSP70 (D) from the same cells. Results for cells treated with heat and/or AUY922 have been normalized for that of the control cells (TID = 0CEM43, AUY922 = 0 nM) in (C) and (D). Data are presented as means ± Std. dev of at least n = 3 independent experiments and statistical significance is denoted with an asterisk ‘⋆‘, and is assumed at p < .05.
Discussion
Sub-ablative heat exerts pleiotropic biological effects on cancers and their characterization could inform clinical decisions e.g., where whole tumor ablation might not be wanted due to targeting feasibility, or when combined with other treatments. Our results show that a sub-ablative TID of 120CEM43 exposure of HCT116 and HT29 colon cancer cells results in
an increase of biomarkers associated with ICD including ATP release, CALR exposure, HSP70, and HSP90 plasma membrane expression, and downregulation of CD47 exposure on the plasma membrane,
synergism with the HSP90 inhibitor AUY922 which decreases the viability of colon cancer cells, and the association of this process with programmed cell death
a further increase of the regulation of pro-immune biomarkers including CALR exposure and CD47 downregulation on the plasma membrane when combined with AUY922, as well as the release of HSP70 when used alone or in combination with AUY922.
We first set out to investigate whether the cytotoxic effect of sub-ablative heat in colon cancer cells () was associated with the induction of ICD biomarkers. As described above, their increase was seen after exposure of both cell lines to a TID of 120CEM43 (). Heating cells with a dose ‘milder’ than 120CEM43 e.g., with a TID of 60CEM43 resulted in less pronounced effects. For example, no CALR exposure 24 and 48 h after treatment and no or negligible levels of ATP release were seen 48 h after treatment in the 2 cell lines. An increase of ATP released from these cells relative to control cells was seen 24 h after treatment () but this may have been associated with the percentage of cells undergoing necrosis and/or apoptosis at that time point [Citation60,Citation62,Citation63]. Hence evidence for the activation of ICD-like mechanisms was stronger in HCT116/HT29 cells exposed to a TID of 120CEM43 than for a TID of 60CEM43, and extend current knowledge which is suggestive of the inability of sub-ablative heat to induce ICD in-vitro [Citation59].
The results presented here also show that the cytotoxic effect of TIDs of 60 and 120CEM43 diminished with time, and that survivors can proliferate and increase their progeny (). Thus to design the most effective treatments there is a need to identify the biological resistance mechanisms exploited by the colon cancer cells [Citation64]. Elegant experiments have shown the importance of the G2/M cell cycle transition phase in the induction of thermotolerance [Citation65]. For this reason, the percentage of heat-exposed HCT116/HT29 cells in the G2/M transition phase of the cell cycle 24 h after treatment was investigated. Results showed that the percentage of 120CEM43-exposed, but not 60CEM43-exposed cells, was increased relative to sham-exposed cells (). In addition, exposure of HCT116 and HT29 cells to a TID of 120CEM43 increased intracellular HSP70, but not HSP90, whereas treatment with a TID of 60CEM43 increased HSP70 expression in HCT116 cells only (). These results show that a pro-survival heat stress response was induced in both colon cancer cell lines exposed to a TID of 120CEM43 possibly mediating the adaptation of heat resistant cells to the thermal stress. The lack of HSP90 regulation can be explained by its over-expressed levels in cancer cells [Citation38], and the role of the 3-dimensional conformation and post-translational modifications in its activation [Citation42]. Since thermal exposure of cells results in protein unfolding and denaturation, and heat shock proteins are associated with their repair [Citation33,Citation66], we sought to find out whether heat shock protein inhibition could enhance, not only cell death but also the release of immunogenic ‘danger’ signals.
To test this hypothesis, heat-exposed cells were treated with the HSP90-specific inhibitor AUY922. Viability data showed that when HCT116 and HT29 cells were exposed to a TID of 120CEM43 in combination with nontoxic concentrations of AUY922 (40 nM), cell viability was reduced in a statistically significant manner, not only relative to sham-exposed cells (no heat or AUY922 treatment) but also compared to those treated with AUY922 or heat alone (). These results also showed that there was increased cytotoxicity only in HCT116 cells exposed to a TID of 60CEM43, which upregulated intracellular HSP70, when treated with AUY922 (). HT29 cells with no upregulated HSP70, when exposed to a TID of 60CEM43, showed no decrease of their viability when combined with AUY922. These results showed the dependence of thermally-treated cells on heat shock proteins for their survival and suggested a potential synergism between the heat and AUY922 treatments. Indeed this was proved after calculating the CI of the combined treatments which were found to be ≪1 for both cell lines (). Furthermore, the lack of synergism between heat and AUY922, for example, in HT29 cells exposed to a TID of 60CEM43 (e.g., lack of statistically significant reduction of viability between 60CEM43 and 60CEM43+AUY922-treated HT29 cells), and in HCT116 and HT29 cells exposed to an ablative TIDs of 240CEM43 () confirmed that these effects may be specific only to a cell type-dependent, relatively ‘narrow’ range of TIDs.
To support the hypotheses set out in these experiments, the effects of the combined heat and AUY922 treatments on PCD were investigated. As expected, an increase of apoptotic biomarkers (e.g., cleaved PARP, cleaved caspase 3, phosphatidylserine (Annexin V) staining) was seen in cells exposed to combined AUY922 and 120CEM43 compared to all other treatments (). These effects were more evident in HCT116 cells which showed a higher loss of viability than HT29 cells. The reason may be that although both HCT116 and HT29 cells absorb AUY922 within 1 h of administration, AUY922 levels are higher in HCT116 than in HT29 cells. This is because HT29 cells express uridine diphosphoglucuronosyltransferases, in contrast to HCT116 cells which do not. These enzymes contribute to higher levels of glucuronidation of AUY922 in HT29 cells than in HCT116 cells, which in turn leads to higher AUY922 levels in HCT116 cells [Citation52]. These differences could account, at least in part, for the greater sensitivity of the HCT116 cells to the combined heat and AUY922 treatment than for the HT29 cells, and further, serve to confirm the dependence of those biological effects on the inhibition of the heat shock response by AUY922.
Advances in cancer immunotherapy have provided clinical benefits for colorectal cancer patients. For example, the development of antibodies that interfere with the function of the CTLA-4 and PD-1 immune regulatory signals (immune checkpoint inhibitor antibodies (ICIs)) has led to significant responses in deficient mismatch repair and microsatellite unstable (high) colorectal cancer patients [Citation67]. However, the low percentage of colorectal cancer patients with these DNA abnormalities means that these treatments only benefit a small percentage of patients, and, as a result, 5-year survival rates remain low (< 15%) [Citation68]. The decrease of immunosuppressive signaling (e.g., CD47 exposure in cancer cells) and the generation of a pro-immune tumor microenvironment (e.g., CALR exposure, HSP70 release) have been postulated as strategies for overcoming this immune resistance. The results of our study showed a statistically significant increase in the percentage of HCT116 and HT29 cells downregulating CD47, and increasing CALR on their plasma membrane after exposure to 120CEM43 and AUY922, relative to all other groups 96 h after treatment. Also, at this time point, only negligible (but statistically significant) increases in CALR exposure, and no down-regulation of CD47 were seen in HCT116 and HT29 cells exposed to heat alone compared to the control cells (). Hence, evidence is shown here that treating colon cancer cells with the combined AUY922 and 120CEM43 gives a longer-lasting pro-immune phenotype to these cells, something that would not otherwise be possible.
HMGB1 is a nuclear chromatin-binding protein involved in DNA architecture and transcriptional regulation [Citation69,Citation70]. It can be released extracellularly in necrotic or injured cells and has been recognized as one of the damage-associated molecular patterns associated with the induction of ICD. After its release from the cell, it can interact with receptors, including the receptor for advanced glycation end products (RAGE), various Toll-like families of receptors (TLRs), and the T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) receptor [Citation71]. These interactions, as well as their oxidation [Citation72], provide the basis for the seemingly opposing roles this protein can have in cancer progression and treatment. On the one hand, it can be required for tumor antigen presentation and the initiation of an anti-cancer immune response after chemotherapy and radiotherapy treatments [Citation73], on the other, there is evidence to suggest its involvement in tumor progression [Citation74–77] and the suppression of nucleic acid-induced anti-tumor immunity [Citation78]. In colorectal cancer, increased HMGB1 concentrations can contribute to the tumor’s growth and metastasis [Citation79–81], and the recruitment of anti-immune regulators (myeloid-derived suppressor cells) upon surgical resection of tumors [Citation82]. In this study, the downregulation of HMGB1 release after treatment with the combined 120CEM43 and AUY922 (40 nM) compared to all other exposures (except heat-exposed HT29 cells) was seen (). The reasons for this regulation remain unclear. Necrosis is not the predominant mode of action the sub-ablative treatments used in this study have. In addition, the active release of HMGB1 from non-necrotic cells requires the initiation of molecular mechanisms, leading to its acetylation and phosphorylation [Citation83,Citation84]. These mechanisms may be compromised by the heat and/or AUY922 treatments, thus leading to an abrogation of HMGB1’s release from cells. In future studies, it would be interesting to investigate whether HMGB1 release, as tested here, could become a prognostic marker for the efficacy of colorectal cancer treatments and/or be associated with improved control of pro-tumoural inflammation in vivo [Citation80,Citation82].
Finally, in this study additional evidence of pro-immune signals came in the form of HSP70 release, another ‘danger’ signal provoking an inflammatory response in the tumor microenvironment [Citation85], after treatment of the HCT116/HT29 cells with 120CEM43 either alone or in combination with AUY922 (). Collectively these results show a pro-immune regulation of ICD biomarkers by sub-ablative heat and/or AUY922 treatments. This makes these treatments potential candidates for use in combination with current checkpoint inhibitor immunotherapies to improve the prognosis of colon cancer patients who do not respond to these treatments. It is not unreasonable to hypothesize that treatment of colon cancer with sub-ablative heat and AUY922, as tested here, could create an immune-stimulatory microenvironment contributing to the recruitment of innate immune cells (e.g., dendritic cells, neutrophils), thus increasing antigen uptake, cell killing, and potentially triggering the adaptive immune response. Checkpoint inhibitor immunotherapy could then maximize the anti-cancer immune response, as has been shown for anti-PD-1 treated, AUY922-sensitised melanoma tumors [Citation44]. The results of this study also contribute to efforts to define the most appropriate thermal doses for inducing an anticancer immune response [Citation86,Citation87], and show that the use of precise sub-ablative TIDs, as shown here and elsewhere [Citation57–59], with well-defined biological and immunological outcomes, can, not only maximize treatment response but also increase the pro-immune phenotype of the cancer cells, thus providing added value when used in combination with the latest immunotherapies.
Before such an outcome can be realized some challenges remain. First, the frequency and magnitude of the thermal treatments have to be fine-tuned in-vivo to facilitate the creation of a pro-immune microenvironment, as tested here in-vitro. Second, the use of immune-competent genetically modified colorectal cancer models would make possible studies that investigate antigen recognition and activation of tumor-specific T cells. Third, the effects of heat and AUY922 on pro-tumoural inflammation need to be investigated in full (e.g., macrophage activation). Fourth, the use of novel HSP90 inhibitors with improved tissue retention properties would maximize the immune stimulatory and cell-killing effects of these treatments.
In summary, this study has shown that exposing HCT116 and HT29 colon cancer cells to cytotoxic, but not ablative, thermal exposures exert a dynamic effect on cell viability, with cytotoxicity being associated with the induction of pro-immune biomarkers 48 h after treatment. However, as soon as 24 h after thermal treatment, some heat resistant cells adapt to the thermal stress by increasing heat shock protein expression, arrest in the G2/M phase of the cell cycle, and then begin to proliferate. Combining heat with targeted therapies against the heat stress response, not only works synergistically to change the fate of these survivors to one of apoptosis-associated cell death but also induce a longer-lasting pro-immune phenotype in at least some of these cells by downregulating CD47 and increasing CALR exposure on their plasma membrane and releasing HSP70. These results show the potential biophysical thermal treatments have to eradicate primary tumors when combining them with molecular oncologic interventions to treat survivor cells outside the ablative focus.
Acknowledgments
The authors would like to acknowledge Prof. Udai Banerji (The Institute of Cancer Research, London, UK) for kindly providing us with the HSP90 inhibitor NVP-AUY922.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Remaining data associated with this study are available in the Supplementary material.
Additional information
Funding
References
- World Health Organization, Global Cancer Observatory, Cancer Today, IARC 69372 Lyon CEDEX08 France; [cited 27 August 2021]. Avaialble from: https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf
- ter Haar G. Therapeutic applications of ultrasound. Prog Biophys Mol Biol. 2007;93(1–3):111–129.
- Meijerink MR, Puijk RS, van Tilborg AAJM, et al. Radiofrequency and microwave ablation compared to systemic chemotherapy and to partial hepatectomy in the treatment of colorectal liver metastases: a systematic review and meta-analysis. Cardiovasc Intervent Radiol. 2018;41(8):1189–1204.
- Marqa MF, Colin P, Nevoux P, et al. Focal laser ablation of prostate cancer: numerical simulation of temperature and damage distribution. Biomed Eng Online. 2011;10:45.
- Vogl TJ, Farshid P, Naguib NN, et al. Thermal ablation of liver metastases from colorectal cancer: radiofrequency, microwave and laser ablation therapies. Radiol Med. 2014;119(7):451–461.
- Illing RO, Kennedy JE, Wu F, et al. The safety and feasibility of extracorporeal high-intensity focused ultrasound (HIFU) for the treatment of liver and kidney tumours in a western population. Br J Cancer. 2005;93(8):890–895.
- Serrone J, Kocaeli H, Douglas Mast T, et al. The potential applications of high-intensity focused ultrasound (HIFU) in vascular neurosurgery. J Clin Neurosci. 2012;19(2):214–221.
- Aptel F, Lafon C. Therapeutic applications of ultrasound in ophthalmology. Int J Hyperthermia. 2012;28(4):405–418.
- Leslie TA, Kennedy JE. High intensity focused ultrasound in the treatment of abdominal and gynaecological diseases. Int J Hyperthermia. 2007;23(2):173–182.
- Van der Kooij SM, Ankum WM, Hehenkamp WJ. Review of nonsurgical/minimally invasive treatments for uterine fibroids. Curr Opin Obstet Gynecol. 2012;24(6):368–375.
- Illing R, Chapman A. The clinical applications of high intensity focused ultrasound in the prostate. Int J Hyperthermia. 2007;23(2):183–191.
- Barqawi AB, Crawford ED. Emerging role of HIFU as a noninvasive ablative method to treat localized prostate cancer. Oncology. 2008;22(2):123–129.
- Li S, Wu PH. Magnetic resonance image-guided versus ultrasound-guided high-intensity focused ultrasound in the treatment of breast cancer. Chin J Cancer. 2013;32(8):441–452.
- Mearini L. High intensity focused ultrasound, liver disease and bridging therapy. World J Gastroenterol. 2013;19:7494–7499.
- Zhou Y. High-intensity focused ultrasound treatment for advanced pancreatic cancer. Gastroenterol Res Pract. 2014;2014:205325.
- Yarmolenko PS, Moon EJ, Landon C, et al. Thresholds for thermal damage to normal tissues: an update. Int J Hyperthermia. 2011;27(4):320–343.
- Dewhirst MW, Viglianti BL, Lora-Michiels M, et al. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int J Hyperthermia. 2003;19(3):267–294.
- Dewhirst MW, Abraham J, Viglianti B. Evolution of thermal dosimetry for application of hyperthermia to treat cancer. Adv Heat Transfer. 2015;47:397–421.
- Wu F. Heat-based tumor ablation: role of the immune response. Adv Exp Med Biol. 2016;880:131–153.
- Garg AD, Dudek-Peric AM, Romano E, et al. Immunogenic cell death. Int J Dev Biol. 2015;59(1–3):131–140.
- Kepp O, Senovilla L, Vitale I, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3(9):e955691.
- Liu P, Zhao L, Kepp O, et al. Quantitation of calreticulin exposure associated with immunogenic cell death. Methods Enzymol. 2020;632:1–13.
- Garg AD, Krysko DV, Verfaillie T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. Embo J. 2012;31(5):1062–1079.
- Fredly H, Ersvaer E, Gjertsen B-T, et al. Immunogenic apoptosis in human acute myeloid leukemia (AML): primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol Rep. 2011;25(6):1549–1556.
- Uscanga-Palomeque AC, Calvillo-Rodríguez KM, Gómez-Morales L, et al. CD47 agonist peptide PKHB1 induces immunogenic cell death in T-cell acute lymphoblastic leukemia cells. Cancer Sci. 2019;110(1):256–268.
- Chao MP, Jaiswal S, Weissman-Tsukamoto R, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94.
- Sapareto S, Dewey W. Thermal dose determination in cancer therapy. Int J Radiat Oncol Biol Phys. 1984;10(6):787–800.
- Sapareto SA. Thermal isoeffect dose: addressing the problem of thermotolerance. Int J Hyperthermia. 1987;3(4):297–305.
- Mouratidis PXE, Rivens I, Civale J, et al. Relationship between thermal dose and cell death for "rapid" ablative and "slow" hyperthermic heating. Int J Hyperthermia. 2019;36(1):229–243.
- Ananthan J, Goldberg AL, Voellmy R. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science. 1986;232(4749):522–524.
- Henle KJ, Leeper DB. Modification of the heat response and thermotolerance by cycloheximide, hydroxyurea and lucanthone in CHO cells. Radiat Res. 1982;90(2):339–347.
- Mizzen LA, Welch WJ. Characterization of the thermotolerant cell. I. Effects on protein synthesis activity and the regulation of heat-shock protein 70 expression. J Cell Biol. 1988;106(4):1105–1116.
- Lepock JR. How do cells respond to their thermal environment? Int J Hyperthermia. 2005;21(8):681–687.
- Burdon RH, Slater A, McMahon M, et al. Hyperthermia and the heat-shock proteins of HeLa cells. Br J Cancer. 1982;45(6):953–963.
- Schumacher RJ, Hansen WJ, Freeman BC, et al. Cooperative action of Hsp70, Hsp90, and DnaJ proteins in protein renaturation . Biochemistry. 1996;35(47):14889–14898.
- Prodromou C, Roe SM, O'Brien R, et al. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90(1):65–75.
- Ritossa F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia. 1962;18(12):571–573.
- Sreedhar AS, Kalmár E, Csermely P, et al. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562(1-3):11–15.
- Wiech H, Buchner J, Zimmermann R, et al. Hsp90 chaperones protein folding in vitro. Nature. 1992;358(6382):169–170.
- Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294.
- Sawarkar R, Sievers C, Paro R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell. 2012;149(4):807–818.
- Kubota H, Yamamoto S, Itoh E, et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones. 2010;15(6):1003–1011.
- Barrott JJ, Haystead TA. Hsp90, an unlikely ally in the war on cancer. Febs J. 2013;280(6):1381–1396.
- Song KH, Oh SJ, Kim S, et al. HSP90A inhibition promotes anti-tumor immunity by reversing multi-modal resistance and stem-like property of immune-refractory tumors. Nat Commun. 2020;11(1):562.
- Huang Q, He S, Tian Y, et al. Hsp90 inhibition destabilizes Ezh2 protein in alloreactive T cells and reduces graft-versus-host disease in mice. Blood. 2017;129(20):2737–2748.
- Banerji U, O'Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23(18):4152–4161.
- Goetz MP, Toft D, Reid J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23(6):1078–1087.
- Weigel BJ, Blaney SM, Reid JM, et al. A phase I study of 17-allylaminogeldanamycin in relapsed/refractory pediatric patients with solid tumors: a children’s oncology group study. Clin Cancer Res. 2007;13(6):1789–1793.
- Egorin MJ, Rosen DM, Wolff JH, et al. Metabolism of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) by murine and human hepatic preparations. Cancer Res. 1998;58(11):2385–2396.
- Kelland LR, Sharp SY, Rogers PM, et al. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst. 1999;91(22):1940–1949.
- Meli M, Pennati M, Curto M, et al. Small-molecule targeting of heat shock protein 90 chaperone function: rational identification of a new anticancer lead. J Med Chem. 2006;49(26):7721–7730.
- Eccles SA, Massey A, Raynaud FI, et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–2860.
- Brough PA, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51(2):196–218.
- Bendell JC, Bauer TM, Lamar R, et al. A phase 2 study of the Hsp90 inhibitor AUY922 as treatment for patients with refractory gastrointestinal stromal tumors. Cancer Invest. 2016;34(6):265–270.
- Seggewiss-Bernhardt R, Bargou RC, Goh YT, et al. Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer. 2015;121(13):2185–2192.
- Piotrowska Z, Costa DB, Oxnard GR, et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann Oncol. 2018;29(10):2092–2097.
- Miyagawa T, Saito H, Minamiya Y, et al. Inhibition of Hsp90 and 70 sensitizes melanoma cells to hyperthermia using ferromagnetic particles with a low curie temperature. Int J Clin Oncol. 2014;19(4):722–730.
- Vriend LEM, van den Tempel N, Oei AL, et al. Boosting the effects of hyperthermia-based anticancer treatments by HSP90 inhibition. Oncotarget. 2017;8(57):97490–97503.
- Adkins I, Sadilkova L, Hradilova N, et al. Severe, but not mild heat-shock treatment induces immunogenic cell death in cancer cells. Oncoimmunology. 2017;6(5):e1311433.
- Mouratidis PX, Rivens I, Ter Haar G. A study of thermal dose-induced autophagy, apoptosis and necroptosis in colon cancer cells. Int J Hyperthermia. 2015;31(5):476–488.
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446.
- Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286.
- Iyer SS, Pulskens WP, Sadler JJ, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106(48):20388–20393.
- Shevtsov M, Balogi Z, Khachatryan W, et al. Membrane-Associated heat shock proteins in oncology: from basic research to new theranostic targets. Cells. 2020;9(5):1263.
- Luft JC, Benjamin IJ, Mestril R, et al. Heat shock factor 1-mediated thermotolerance prevents cell death and results in G2/M cell cycle arrest. Cell Stress Chaper. 2001;6(4):326–336.
- Roti Roti JL, Kampinga HH, Malyapa RS, et al. Nuclear matrix as a target for hyperthermic killing of cancer cells. Cell Stress Chaper. 1998;3(4):245–255.
- Breakstone R. Colon cancer and immunotherapy-can we go beyond microsatellite instability? Transl Gastroenterol Hepatol. 2021;6:12.
- Zhang Y, Rajput A, Jin N, et al. Mechanisms of immunosuppression in colorectal cancer. Cancers. 2020;12(12):3850.
- Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins . Mol Cell Biol. 1999;19(8):5237–5246.
- Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 2003;22(20):5551–5560.
- Klune JR, Dhupar R, Cardinal J, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14(7–8):476–484.
- Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860–875.
- Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059.
- Jube S, Rivera ZS, Bianchi ME, et al. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72(13):3290–3301.
- Palumbo R, Sampaolesi M, De Marchis F, et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164(3):441–449.
- Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405(6784):354–360.
- Yang GL, Zhang LH, Bo JJ, et al. Increased expression of HMGB1 is associated with poor prognosis in human bladder cancer. J Surg Oncol. 2012;106(1):57–61.
- Chiba S, Baghdadi M, Akiba H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–842.
- Ueda M, Takahashi Y, Shinden Y, et al. Prognostic significance of high mobility group box 1 (HMGB1) expression in patients with colorectal cancer. Anticancer Res. 2014;34(10):5357–5362.
- Zhang Z, Wang M, Zhou L, et al. Increased HMGB1 and cleaved caspase-3 stimulate the proliferation of tumor cells and are correlated with the poor prognosis in colorectal cancer. J Exp Clin Cancer Res. 2015;34(1):51.
- Huang M, Geng Y, Deng Q, et al. Translationally controlled tumor protein affects colorectal cancer metastasis through the high mobility group box 1-dependent pathway. Int J Oncol. 2018;53(4):1481–1492.
- Li W, Wu K, Zhao E, et al. HMGB1 recruits myeloid derived suppressor cells to promote peritoneal dissemination of Colon cancer after resection. Biochem Biophys Res Commun. 2013;436(2):156–161.
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195.
- Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177(11):7889–7897.
- Calderwood SK, Theriault JR, Gong J. Message in a bottle: role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur J Immunol. 2005;35(9):2518–2527.
- Qu S, Worlikar T, Felsted AE, et al. Non-thermal histotripsy tumor ablation promotes abscopal immune responses that enhance cancer immunotherapy. J Immunother Cancer. 2020;8(1):e000200.
- Fite BZ, Wang J, Kare AJ, et al. Immune modulation resulting from MR-guided high intensity focused ultrasound in a model of murine breast cancer. Sci Rep. 2021;11(1):927.