
ABSTRACT
The Aerosol Mass Spectrometer (AMS) and Aerosol Chemical Speciation Monitor (ACSM) are widely used for quantifying submicron aerosol mass concentration and composition, in particular for organic aerosols (OA). Using the standard vaporizer (SV) installed in almost all commercial instruments, a collection efficiency (CE) correction, varying with aerosol phase and chemical composition, is needed to account for particle bounce losses. Recently, a new “capture vaporizer” (CV) has been shown to achieve CE∼1 for ambient aerosols, but its chemical detection properties show some differences from the SV due to the increased residence time of particles and vaporized molecules inside the CV. This study reports on the properties and changes of mass spectra of OA in CV-AMS using both AMS and ACSM for the first time. Compared with SV spectra, larger molecular-weight fragments tend to shift toward smaller ions in the CV due to additional thermal decomposition arising from increased residence time and hot surface collisions. Artifact CO+ ions (and to a lesser extent, H2O+), when sampling long chain alkane/alkene-like OA (e.g., squalene) in the CV during the laboratory studies, are observed, probably caused by chemical reactions between sampled OA and molybdenum oxides on the vaporizer surfaces (with the carbon derived from the incident OA). No evidence for such CO+ enhancement is observed for ambient OA. Tracer ion marker fractions (fm/z =, i.e., the ratio of the organic signal at a given m/z to the total OA signal), which are used to characterize the impact of different sources are still present and usable in the CV. A public, web-based spectral database for mass spectra from CV-AMS has been established.
Copyright © 2018 American Association for Aerosol Research
EDITOR:
1. Introduction
Organic aerosol (OA) accounts for 10–90% of the total submicron aerosols in the ambient air (Kanakidou et al. Citation2005; Murphy et al. Citation2006; Zhang et al. Citation2007; Jimenez et al. Citation2009) and hence has a substantial impact on climate forcing (Poschl, Citation2005; IPCC, Citation2017) and human heath (Davidson et al. Citation2005; Mauderly and Chow Citation2008). The quantification and chemical speciation of OA (comprised of thousands of individual molecular species) are always challenging due to its complex properties. In recent years, Aerosol Mass Spectrometer (AMS) and Aerosol Chemical Speciation Monitor (ACSM), commercialized by Aerosol Research, Inc. (ARI), have been widely used to quantify the mass concentration and size distributions of submicron OA at a time resolution of minutes or faster (Allan et al. Citation2003a; Drewnick et al. Citation2004; Drewnick et al. Citation2005; DeCarlo et al. Citation2006; Canagaratna et al. Citation2007, and references therein).
A substantial fraction of the quantification uncertainty of AMS/ACSM instruments is dominated by the collection efficiency (CE), mainly due to particle bounce on the surface of the standard vaporizer (hereinafter referred to as “SV”) installed in almost all currently operating instruments (Matthew et al. Citation2008; Middlebrook et al. Citation2012). CE varies from ∼0.45 – 1 for ambient aerosols (typically ∼0.5), with lower values possible for laboratory aerosols. Particle bounce can also influence the detected fragmentation and size distributions of aerosols in AMSs/ACSMs, which complicates interpretation of AMS measurements (Liao et al. Citation2017; Robinson et al., Citation2017). A new “capture vaporizer” (CV) has been recently developed to reduce particle bounce fraction (Jayne and Worsnop Citation2016). While the SV has an inverted cone shape and a porous tungsten surface, the CV was designed to have a “cage” with a narrow entrance and an internal structure favoring repeated internal bounce, in order to increase the residence time of particles in contract with the hot vaporizer surfaces, and, thus, reduce the fraction of particles that bounce without evaporation. The CV is made of solid molybdenum, in contrast to the porous tungsten used for the SV. The change of material for the CV was due to the higher feasibility of manufacturing the more complex geometry of CV (Figure S1a) using molybdenum than porous tungsten. We note that molybdenum was used in very early AMS studies (Allan et al. Citation2003b; Jimenez et al. Citation2003). To decrease the particle bounce fraction, the SV was developed using porous tungsten. Other materials like nichrome (90% Ni/10% Cr) (Voisin et al. Citation2003) and a copper rod coated with a fluorinated grease (Tobias et al. Citation2000) have also been used to collect and thermally desorb particles in other aerosol instruments, however heated to lower temperature (below 200°C).
The quantification, fragmentation, and size distribution of inorganic aerosols in the CV have been described in detail for laboratory studies (Hu et al. Citation2017b; Xu et al. Citation2017) and field studies (Hu et al. Citation2017a). A CE of ∼1 for ambient total and inorganic aerosols are observed in AMS/ACSM measurements with the CV, suggesting the elimination of particle bounce in the CV for internally mixed ambient aerosols (Hu et al. Citation2017a). Bounce is significantly reduced, but not always eliminated, for pure inorganic species in the laboratory, which is qualitatively consistent with the stronger tendency of some pure particles to bounce in the SV, compared to ambient particles. With longer residence time of aerosols in the CV, the larger inorganic (e.g., sulfate and nitrate) molecular-weight ions tend to shift to smaller ions, due to additional thermal decomposition processes.
The changes in the chemical detection of OA with the CV has not yet been studied. In particular it is important to clarify if a CV-AMS can provide as much useful chemical information on OA as a SV-AMS can. It is also important to document whether the ion markers that are well established for SV-AMS are still useful for the CV-AMS. To investigate those questions, we have analyzed various types of OA from different laboratory, chamber, and field studies, sampled by two co-located AMSs (or two ACSMs) equipped with the SV and CV, respectively. For reference, the abbreviations and acronyms used in this study are explained in Table S1.
2. Experiment and instrumentation
2.1. Laboratory, chamber, and field experiments
The setup of laboratory, chamber, and field experiments for this study is shown in Figure S1b-f. In the laboratory experiments, three aerosol generation methods for pure standard OA species were used:
| (i) | Atomization method: OA was produced by running dilute solutions into a Collison atomizer (model: 3076; TSI, US), and a silica gel diffusion dryer was used to remove the solvent prior to chemical analysis in order to reduce interferences. Isopropanol or hexane were used as solvents for squalene (C30H50), oleic acid (C18H34O2), and octacosane (C28H58). Water was used for most of the water soluble compounds, e.g., highly oxidized acids. | ||||
| (ii) | Laser ablation: OA in pure liquid phase or in solutions was first dropped on a cleaned, thin-layer chromatography (TLC) or glass plate. Then, OA particles were generated through irradiation of the solid material with a laser beam (YAG laser, 213 nm), generated by a commercial laser ablation system (LSX 213, CETAC Technologies, Omaha, NE, USA) (Gunther and A. Heinrich Citation1999; Reifschneider et al. Citation2015). The laser operating conditions, which determine the particle size and mass production rate, can be controlled by adjusting the laser spot size (4–200 μm), intensity (5–20% of full energy of 4 mJ), and scan speed (50 µm s−1). An advantage of this method is that only a small amount of OA mass is needed, and the solvent is completely removed prior to chemical detection. | ||||
| (iii) | Evaporation-condensation apparatus: Dioctyl sebacate (DOS, C26H50O4) particles were generated based on vapor re-condensation by mixing of cooler air after being evaporated in a heated glass flask (Aimanant and Ziemann Citation2013). Then, the DOS particles were flushed into a clean 20 m3 Teflon chamber in the CU Atmospheric Chamber Facility (Krechmer et al. Citation2017) and sampled. DOS was the only OA species generated by method (iii), and was also used as OA seed for secondary OA (SOA) formation experiments in the chamber. | ||||
The detailed information for the generation methods, gas carrier and solvent used for different pure compounds in this study is summarized in Table S2. For the chamber studies, SOA from oxidation of α-pinene and Δ-carene with NO3 radicals in the same chamber were sampled with the CV- and SV-AMSs. SOA from ozone-initiated oxidation of α-pinene in the chamber was also analyzed in the CV for this study.
Data from three field studies were also analyzed to compare the performance of the SV and CV: Southern Oxidation Aerosol Study (SOAS) (Carlton et al. Citation2018); sampling of ambient air in Billerica, MA (Hu et al. Citation2017a); and the KORean-United States Air Quality mission (KORUS-AQ, https://espo.nasa.gov/home/korus-aq). The SOAS study was conducted at a ground site in a pollution-influenced forest area in the Southeastern US (Centreville, AL, USA) during the summer of 2013 (Carlton et al. Citation2018). The ambient OA in this study was dominated by biogenic sources, mainly oxidation products from isoprene (Hu et al. Citation2015; Krechmer et al. Citation2015) and monoterpenes (Xu et al. Citation2014), with smaller contributions from other sources (Kim et al. Citation2015). The KORUS-AQ study (May-June, 2016) was an aircraft-based field campaign to investigate the factors controlling air quality in and around the Seoul Metropolitan Area; anthropogenic (vehicular and industrial) sources strongly influence this campaign. Some impact from aged long-range transport sources was also expected. The Billerica study (September, 2012) was performed by sampling ambient air on the campus of Aerodyne Research Inc., located in a suburb of Boston, MA (USA).
2.2. AMS/ACSM
For the laboratory and chamber studies, two high-resolution time-of-flight AMSs (HR-ToF-AMS, DeCarlo et al. Citation2006), equipped with an SV or CV, were deployed, as previously described in Hu et al. (Citation2017a,b). In the SOAS study, an HR-ToF-AMS with the SV was used for the entire campaign, while a compact time-of-flight AMS (C-ToF-AMS, Drewnick et al. Citation2005) coupled with the CV was used for most of the campaign. To obtain high resolution spectra data in the CV measurement, we changed the C-ToF detector to an HR-ToF detector with the same setup of the rest AMS body during a few days in SOAS (7—10 June). For the Billerica study, two ACSMs (Ng et al. Citation2011) were used. In KORUS-AQ, two HR-ToF-AMSs were installed on the NASA DC-8, but sampled through different inlets. The detailed setups of the AMS sampling can be seen in Figure S1. The AMS inlet line relative humidity (RH) was always under 35% for ground campaigns and 45% for aircraft measurement; so, the aerosols had low water content. An exception is the Billerica study, where no dryer was used. Details on the calibration protocol of the AMS are reported in Hu et al. (Citation2017a,Citationb).
3. Results and discussion
3.1. OA spectra comparison of the SV vs. CV
Compared to the spectra obtained with the SV, the organic ion distributions of the CV are generally shifted towards smaller m/z ( and S2–3), for pure standards, chamber SOA, or ambient OA. The normalized difference spectra (CV minus SV divided by SV) show negative values for the majority of ions above m/z 44 ( and S3). The fraction of signal at m/z < 50 is substantially larger for the CV compared with that in the SV (up to a factor of 2) across a wide range vaporizer temperature range (Tv = 200–800°C), as shown in Figure S4. This shift is found for all OA types and oxidation levels, and is thought to be due to additional thermal decomposition or internal energy resulting from the multiple collisions and longer residence time of aerosols in the CV, and similar to the behavior of inorganic species (Hu et al. Citation2017a,b; Xu et al. Citation2017).
Figure 1. Spectra and comparisons of different pure standard organic compounds: (a) squalene, (b) oleic acid, (c) citric acid, and (d) levoglucosan, between the SV and CV. (e) Chamber SOA generated from NO3+α-pinene with ammonium sulfate (AS) seed, and (f) ambient OA during the SOAS study, between the SV and CV. The top and middle rows are the spectra measured in the SV-AMS and CV-AMS, respectively. The bottom row is the ratio of CV minus SV divided by SV. All the spectra are shown as stacked at unit mass resolution, although they were fit with high-resolution fitting. Only ions contributing more than 1% of total OA to the SV are shown in the differential plot. For squalene, oleic acid, and citric acid, the interference of isopropanol (C3H8O) solvent is less than 5% of total signal and was excluded based on the ratios between the characteristic ion m/z 45 (C3H5O+) and other ions in the pure isopropanol AMS spectrum. Organic H2O+ and CO+ ions from standard compounds were directly calculated using HR-fitting. Organic H2O+ and CO+ ions in chamber SOA were directly calculated using HR fitting. Organic H2O+ ions in the SOAS study was constrained based on organic CO2+ (H2O+/CO2+ = 0.225). Organic CO+ in the ambient CV dataset were calculated based on the enhanced HR-fitted CO+ versus total OA during the OA plumes. Canagaratna et al. (Citation2015) showed that spectral differences among three different SV-AMS instruments were minor.
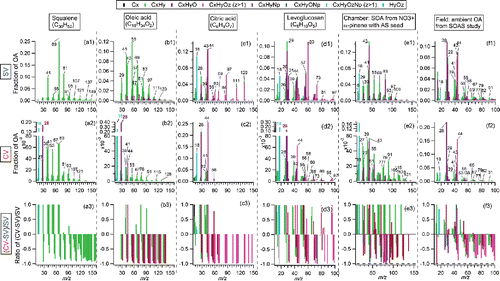
Ratios of different types of ions (e.g., CxHy+; CxHyO+) for three m/z ranges between the CV and SV are summarized in . Enhanced abundance of CxHy+ ions at m/z < 50 are observed for all types of OA measured for the CV, except oleic acid. The low CxHy+ ratios of oleic acid may be related to the unexpected increase of CO+ peak in the CV, which will be discussed in Section 3.2.
Figure 2. Summary of direct ion signal ratios between the CV and SV (CV versus SV) for different OA species. Different types of ions (i.e., CxHy+; CxHyO+; CxHyOz+, CxHyN+, CxHyON+) in the spectra at three m/z ranges (i.e., m/z < 50; m/z = 50–100; m/z > 100). To minimize the uncertainties, only ions with abundance in total OA above 1% in the SV spectrum are included in the calculation. Thus, prominent CO+ ion shown in the squalene spectrum measured by the CV is not taken into account due to its low abundance in the spectrum of the SV (<1%). The arrows in the graph indicate the bar on its left side are above the maximum Y values. All the ambient data were obtained in V-mode in AMS, thus the nitrogen-containing ions are hard to be separated. W mode data were used in chamber studies.
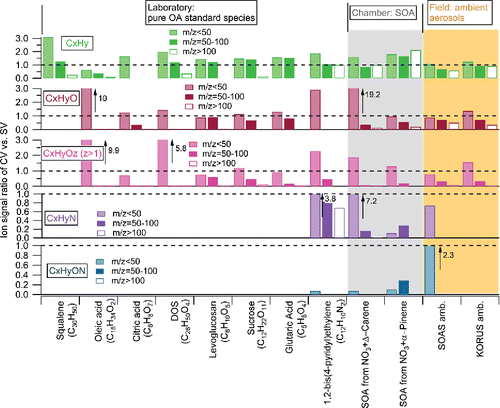
The differences in the spectra ( and S3) suggest that increased thermal decomposition affects the CxHyO+ and CxHyOz+ (z > 1) ions more than the CxHy+ ions in the CV. Similarly, shows almost all the CxHyO+ and CxHyOz+ (z > 1) ratios (CV/SV) at m/z > 50 (except DOS) are lower than the corresponding CxHy+ ratios. More impact of thermal decomposition on CxHyO+ and CxHyOz+ (z > 1) ions than CxHy+ is expected since thermal decomposition reactions are more favorable for the oxygen containing compounds (e.g., acid and alcohol) than pure hydrocarbon compound (Moldoveanu Citation2009). Consistent with that, higher abundance of CO2+, CO+, and H2O+ ions (which are products of de-functionalization reactions) were observed in the CV than SV in all cases ( and S3-S4). At m/z > 100, the ambient OA exhibits higher CV vs SV ratios from CxHyO+ and CxHyOz+ (z > 1) ions than chamber SOA and pure standard OA (), suggesting less additional thermal decomposition effect for ambient aerosols than others. The reason for higher abundances of CxHyO+ and CxHyOz+ (z > 1) ions in ambient OA is not clear, although it may be related to the higher fragility of ambient OA, which already leads to extensive thermal decomposition in the SV (thus reducing the relative impact of additional decomposition in the CV). The fact that complex ambient OA tends to preserve more chemical information at high m/z in the CV is likely to be beneficial for source identification analysis for ambient OA. The N-containing hydrocarbon ions (CxHyN+ and CxHyON+) ratio showed similar changes as CxHy+ ions, however did not show a consistent variation pattern among limited different samples.
Tuning the lens position to focus on the edge of the CV (rather than the center) was shown to lead to mass spectra much more similar to those in the SV for inorganic species, presumably due to reduced exposure of the particles and vapors to the high vaporizer temperatures (Hu et al. Citation2017a). The same phenomenon is observed for OA spectrum (e.g., citric acid), with higher abundance of larger ions (Figure S5), essentially resulting in similar spectra as in the SV. As changing the lens position is far easier than replacing the vaporizer, this technique may have some applicability in practice for larger particles (>150 nm, as particles smaller than 150 nm are not tightly focused by the aerodynamic lens), in particular when trying to compare results of CV studies with those of literature SV studies.
3.2. Causes for extra CO+ and H2O+ production
As shown in the OA spectra comparison in and S3, unusually high abundance of CO+ (and to a lesser extent, H2O+) in the spectra of CV was observed from some laboratory-generated chemically-reduced OA particles.
Atomized long-chain alkene (squalene, C30H50) and alkane particles (octacosane, C28H58, Figure S6a), which do not contain any oxygen in the compounds, show unexpectedly high abundance of CO+ (fCO > 0.15; where fCO = CO+/total OA signal) and H2O+ (squalene: fH2O = 0.13) in their spectra in the CV but not in the SV. Large CO+ and H2O+ enhancements in the CV (fCO > 0.3 and fH2O > 0.2) were also observed from oleic acid (C18H34O2, nominal O:C = 0.11), while elevated but smaller fCO (∼0.06) and fCO2 (∼0.05) were found for 1,2-bis(4-pyridyl)ethylene (C12H10N2). For chamber SOA from α-pinene+NO3 (O:C ≈ 0.2–0.6, including several seed/seedless cases) and Δ-Carene+NO3 (O:C ≈ 0.4), CO+ and H2O+ are higher for the CV than the SV. However, anomalously high CO+ and H2O+ peaks were not observed for highly-oxidized, citric acid (C6H8O7; O:C ≈ 1.2) or chamber SOA of α-pinene + O3 (O:C ≈ 0.8, Figure S6).
Importantly, no additional CO+ enhancement in the CV was observed for the ambient OA. This is true whether the OA originated from biogenic emission dominated environments during the SOAS study (OA ranges from <0.1 to 15 µg m−3, O:C ranges from 0.5–1.0) or urban emission dominated environments in the KORUS-AQ study (OA exceeding 30 µg m−3, O:C ranges from 0.4–1.3). We measured fCO based on the enhanced CO+ and CO2+ and OA above the background signals when high concentration OA plumes were sampled. An example of the measurement of fCO in these ambient datasets is shown in Figure S7. Similar organic fCO (0.17–0.19) and fCO2 ratio (0.15) in the CV from KORUS-AQ are found, as shown in Figure S8a. Similarly, consistent fCO (0.09) vs fCO2 (0.09) in the SV (Figure S8b) was also found. The fCO/fCO2 ratio in the CV (1.13) is comparable or slightly higher than that of the SV (1.05). With similar method, the fCO/fCO2 of CV in the SOAS study was determined as 1.47 (vs 1.35 of SV, ). The default fCO/fCO2 in the fragmentation table of PIKA analysis software for AMS is around 1 (Aiken et al. Citation2008). The abundance of H2O+ from ambient OA is very difficult to quantify due to the potential interferences from ambient gas and particulate water or other species. Thus, ambient H2O+ from OA is conventionally constrained based on the intensity of organic CO2+ ion (Aiken et al. Citation2008). The H2O+/CO2+ ratios in the pure highly oxidized acids, closer in composition to ambient SOA, show comparable or even lower values from the CV (0.5 for citric acid and 1.4 or glutaric acid) than from the SV (0.8 and 1.5, respectively), suggesting that the anomalous enhancement of H2O+ might also not be significant for ambient OA in the CV.
The CO+ and H2O+ enhancements in the CV for long-chain alkene/alkane like standards are surprising. It is necessary to explore the possible reason for this extra CO+ and H2O+ ion production further in order to understand, reduce, or correct for such contamination in the future experiments, such as done for artifact CO2 formation (Pieber et al. Citation2016). Here we use three standard species, squalene (C30H50), oleic acid (C18H34O2) and citric acid (C6H8O7), as examples across a wide range of oxygen content to investigate the possible reason for such extra CO+ and H2O+ production in the CV.
Among all the factors that could potentially lead to CO+ and H2O+ enhancements, we first check if the CO+ ion comes from air or an oxygen-containing solvents (isopropanol, C3H8O). After changing the carrier gas from air to argon (Ar) (O2 is less than 1% of total Ar signal) and isopropanol to hexane (C6H14, >95%), the high fCO (>0.15) and fH2O (>0.2) were still observed in the spectra of oleic acid ( and ) and squalene (Figures S9b and c). Also, the fCO and fH2O values in oleic acid and squalene do not show regular variation patterns with different carrier gas and solvent (Figure S10). Laser-ablation particle generation can exclude the potential inferences from the solvent. The oleic acid and squalene particles formed through the laser ablation system with Ar, as a carrier gas, show similar high fCO and fH2O in their spectra using the CV ( and S10). The consistent appearance of enhanced CO+ and H2O+ ions in the CV spectra (with some experiment-to-experiment variability) suggested that the unexpected CO+ and H2O+ are largely not due to the aerosol generation system, solvent, and/or carrier gas. Therefore, the enhancement must come from the OA itself and/or products formed through interaction between OA and the CV.
Figure 3. (a)-(d) Spectra of normal oleic acid from different aerosol generation systems. (e) The calculated spectrum of oleic acid spectra by assuming all carbons to be isotopically- labeled carbons (13C) based on the measured spectrum in (d). (f) Measured spectrum of isotope 13C labeled oleic acid.
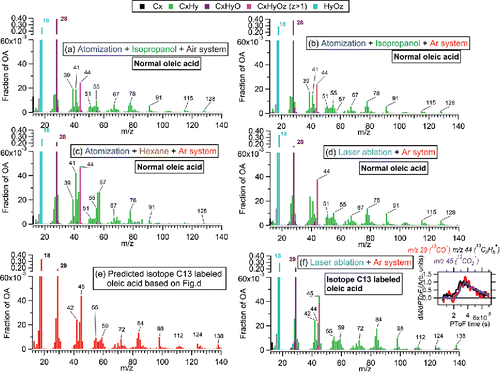
To evaluate this possibility, we used isotopically-labeled oleic acid (13C18H34O2, MW = 300; purity >98%, hereinafter referred to as oleic13) with all 18 carbon atoms (12C) being replaced with 13C. Pure oleic13 particles were generated using the laser ablation system with Ar as carrier gas. If the carbon of the CO+ ion comes from oleic acid itself, then the CO+ ion should move from m/z 28 to m/z 29, while other C-containing ions will shift toward higher ion masses accordingly, based on the number of carbons in the ions. The measured spectrum of oleic13 in the CV () shows very good consistency with the predicted spectrum based on the spectrum of normal oleic acid, as shown in . In the measured spectrum, almost all the CO+ signal (>99%) shifts from m/z 28 to m/z 29 (13CO+), providing that the carbon in CO+ indeed comes from oleic acid. The consistent particle time-of-flight (PToF) distributions between 13CO+ and other ions (e.g., 13CO2+ and 13C3H5+, as shown in the inset of ) also verifies that the CO+ ion is formed rapidly through interaction process between oleic acid and the vaporizer material or impurities/residues. In addition, similar PToF distributions of CO+ and other CxHy+ ions are found for squalene as well. Although H2O+ ions do not contain isotopic carbon, the PToF distributions indeed suggest enhanced H2O+ ions from squalene in PToF and oleic acid particles are also formed rapidly from particle material (not shown).
Since squalene and octacosane do not contain oxygen, we speculate that the oxygen in the CO+ and H2O+ ion may come from oxides on the vaporizer surface, or possibly from particle residues deposited on the vaporizer. Since oleic acid contains oxygen, part of the oxygen in the CO+ and H2O+ ions might be from the compound itself, similar to the CO2+ ion. Unfortunately, we were not able to locate any commercially 18O-labeled oleic acid. We have investigated the variation of CO+, H2O+, CO2+, and other hydrocarbon ions from different OA species using particle beam modulation experiments (Drewnick et al. Citation2015; Hu et al. Citation2017b), as an indirect tool to explore possible origins of CO+ and H2O+ ions, as shown in .
Figure 4. Results of an experiment slowly alternating beam-open and beam-closed positions while sampling (a) squalene, (b) oleic acid, and (c) citric acid in the CV and (d) citric acid in the SV under vaporizer temperature of 550–600°C. The time interval between datapoints is 4–5 s. τ is the lifetime of signal decay and was estimated with an exponential fit to the relevant part of the time series. An example of exponential fitting line for τ is shown in (a). τ for the rising signal is not shown, since it always varies in the same way as τ for the decay signal. Grey background in graph indicates periods when particle beam is blocked, the white background shows the period when particle beam is open and reaching the vaporizer.
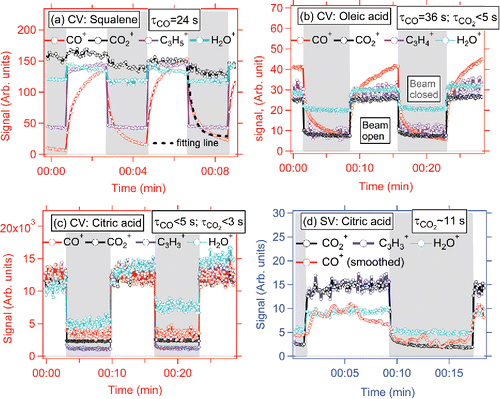
During typical MS mode operation in the AMS, the beam-open (chopper open) and beam-blocked (chopper closed) positions are alternated every several seconds (usually ∼5 s). During particle beam modulation experiments, to study the signal response from short to much longer timescales, the intervals were extended to 5–10 min each. The time resolution of spectra collection/averaging time used was 4–5 s in this study. The decay time, τ, is defined as the e-fold lifetime of signal decay when closing the particle beam after a long period of exposure to incoming particles. τ from different OA species was estimated through an exponential fit to the relevant part of the signal time series. An example of fitting is demonstrated in .
Much slower rise and fall of the CO+ ion from squalene and oleic acid was observed (τ = 24 s and 36 s, respectively), compared to their CxHy+ and CO2+ ions (<5 s). This suggests some CO+ arises from a slower chemical interaction between OA and/or vaporized gases and vaporizer surfaces; thus, resulting in the slow evolution of CO+ but not of CO2+ (note however that some of the CO+ evolution is very fast, as evidenced by the PToF data described above; PToF has a millisecond resolution, which is quite fast compared to second level in MS mode). As a comparison, CO+ from citric acid (which is not enhanced in the CV) shows similar τ as for CO2+ and CxHy+ ions. Since citric acid has multiple functional groups (-carboxylic acid and alcohol groups), it is prone to experience thermal defunctionalization processes, resulting in high CO+ and CO2+ ion abundances in the spectrum, even in the SV. The similarly fast decay of CO+ and CO2+ suggests the oxygen in the CO+ ion of citric acid particles originate directly from prompt vaporization/decomposition in the CV.
Figure S4 shows the fCO as a function of vaporizer temperature (Tv) from squalene, oleic acid, and citric acid. The fCO for squalene and oleic acid depends on Tv, while less so for citric acid. The optimum Tv for producing CO+ from squalene particles is around 500–700°C, and >500°C for oleic acid particles, suggesting that the artificial CO+ ion formation is caused by chemical reactions that are sensitive to temperature. This Tv dependent fCO trend has contributions from the absolute amount of CO+ mass formed at different Tv, and also from faster CO+ and CO2+ signal rise/decay at higher Tv (Figure S12), which will results in higher signals in the difference mode (signal – background with 5 s intervals). The reasons why faster rise/decay can result in higher difference signals and how beam interval can affect the difference signal are demonstrated in the Section 1 and Figure S13 in the supporting information (Salcedo et al. Citation2010). Slight changes of difference and background CO+ signal as a function of time were found in the CV under Tv of 500–600°C, which were different for different species (Figure S14). This suggests that the instrumental history may have a minor influences on the fCO ratio in the CV. It is unclear at this point that whether the enhanced CO+ in the CV is due to the different material (Mo vs. W) or to the CV geometry. Future experiments are needed to address this issue.
Although we cannot experimentally determine the origin of O from CO+ ions, a hypothesis can be formulated based on the literature. Previous studies report that although Molybdenum (Mo) (of which the CV is made) has many useful mechanical properties, it does not have a strong resistance to the oxidation (Gulbransen et al. Citation1963). Mo can be easily oxidized to MoOx (MoO3, and to a lesser extent MoO2) in the presence of O2. MoO3 has been shown to serve as a catalyst and supply oxygen for the oxidation of C4 hydrocarbons to CO/CO2 (Ozkan et al. Citation1990b). In those literature studies (conducted a higher pressures) CO2 was formed in higher abundance than CO. We speculate that the vacuum conditions of our study limit the conversion of CO into CO2. The O in the CO+ may be supplied by lattice oxygen trapped previously in MoO3 (Ozkan et al., Citation1990a).
MoO3 formation and volatilization is temperature-dependent (Gulbransen et al., Citation1963). For temperatures below 450ºC, Mo oxides can be formed, however, adhesion of Mo oxides films to the bulk Mo is very tight, and the surface oxides are not very chemically/catalytically active. For temperatures between 500–700ºC, Mo oxide are less tightly bound and are chemically active. For temperatures above 800°C, liquid Mo oxides can form, however they volatilize as soon as they form, which decreases the catalytic efficiency. The artificial fCO enhancement in the AMS, with maximum at 500–750°C from squalene/oleic acid in CV (Figures S4g and h), is consistent with the optimum catalytic temperature for Mo oxides, supporting this hypothesis. CO2 can be produced on the surface of tungsten-made SV while sampling inorganic particles (Pieber et al. Citation2016), which is conceptually similar to the phenomenon described here.
Unlike CO+, H2O+ shows fast decay (<5 s), similar to hydrocarbon ions (<5 s), for all the compounds, implying the production processes for excess H2O+ in the CV might be different than for CO+. Differences between the evolutions of H2O+/OA and CO+/OA vs. Tv (Figure S4) also support this hypothesis. Based on hypothesis of MoO3 catalytic reactions proposed for the CO+ formation (with C provided by the aerosol and O by the vaporizer), it is plausible that hydrogen from organic aerosol compounds can be oxidized to be H2O. However, a different reaction dynamics than for CO+ formation is apparent, since their decay times, peak temperatures and signal background trend with time are different. Testing with, e.g., deuterated oleic acid (which was not available during our experimental period) might improve the understanding of artifact H2O+ formation in the CV.
In summary, attention should be paid to the enhancement of CO+ and H2O+ in the spectra of long-chain alkene/alkane or similar OA in the CV in future laboratory studies, while this effect appears to be much less important for the more oxidized ambient aerosols. This difference may be due to oxidized species readily undergoing prompt thermal decomposition on the vaporizer, while alkanes and alkenes do not promptly decompose, and thus can take part in slower catalytic reactions on the CV surface. Particle beam modulation experiments (with extended chopper interval) are a useful method to investigate this issue. Exploration of other capture vaporizer materials that may reduce this CO+ artifact should be done in future studies.
In almost all AMS/ACSM studies, CO+ and H2O+ are estimated from CO2+ due to the difficulties in their direct measurement (Aiken et al. Citation2008; Canagaratna et al. Citation2015). If these ions are actually measured and included into the OA concentration for cases in which artificially high signal is present, this effect could lead to artificially higher relative ionization efficiency of OA (RIEOA). This would be due to the added O (since the C does appear to come from the analyzed compound). In the more common case in which CO+ and H2O+ are estimated from CO2+ that may lead to lower RIEOA due to missing some of the C in the compound. These impacts on quantification need to be investigated in future studies. Due to the high degree of species to species variability in RIEOA, it is generally recommended that RIEOA be measured for laboratory species of interest before calculating their mass concentrations (Jimenez et al. Citation2016). Since CV spectra of long-chain alkene/alkanes show significant differences between the estimated and measured intensities of CO+ and H2O+, care must be especially taken to calculate RIEOA values with the same fragmentation wave that is used in the mass concentration calculations for these species. Since no artificial CO+ enhancement has been observed in ambient OAs detected by the CV (at least within the scope of our study) and H2O+ ion has always been constrained based on measured organic CO2+ ion, after proper calibration, the quantification of OA and OA elemental ratios from field studies should not be biased due to this effect. A possible exception is the detection of reduced ambient species such as hydrocarbon-like OA (HOA), which was not abundant for the field studies investigated here.
3.3. Comparison of characteristic ions
In previous sections, we compared the average ion abundance between the SV and CV. Here, the dynamic covariance of important tracer ions between two vaporizers is further examined. Tracer ions in AMS spectra have been shown to be very useful for identifying the impacts from OA sources, and some ions can be used as real-time source tracers. We examine the changes in some of the ions from the SV to CV in this section.
In AMS spectra, m/z 44 (CO2+), mainly formed through thermal decarboxylation of oxo-, di- and poly-carboxylic acids, is a marker for organic acids and a good indicator of OA aging (Alfarra et al. Citation2004; Ng et al. Citation2010; Duplissy et al. Citation2011). Significantly higher f44 values (due to enhanced thermal decomposition/decarboxylation processes) is found for the CV across most laboratory and field studies, as shown in . This enhancement (∼2 on average, with a range of 1–4) varies with different types of OA and degree of oxidation. The correlation indicates that although the absolute values of f44 differ between CV and SV, the trends in f44 are similar across species, indicating that differences in f44 can still be used as an indicator of OA.
Figure 5. Comparison of (a) f44 and (b) f43 between the CV and SV during various laboratory, chamber, and ambient studies. Scatter plots of f44 vs f43 from the (b) CV and (d) SV. Also shown are triangle lines for the SV obtained from Ng et al. (Citation2010) as well as our proposed triangle lines for ambient data only, updated for the CV to guide the eye. The equation for the left line of triangle is estimated to be f44 = −8.148 × f43+0.445, and right line is f44 = −1.964 × f43 + 0.396. KORUS data from research flight 12 (24-May-2016) and 18 (04-June-2016) were used here.
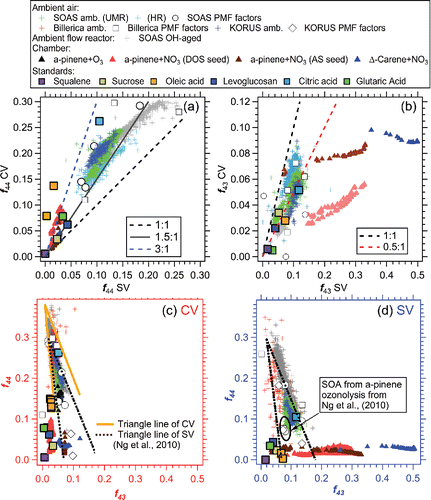
Differences of f44 (up to 40%) have been reported across multiple SV ACSM/AMSs when sampling the same ambient aerosols (Crenn et al. Citation2015; Fröhlich et al. Citation2015). This variability of f44 in the SV is thought to be mainly caused by (1) the presence of a slowly decaying CO2+ signal in some instruments and/or (2) interference of artificial CO2+ produced by interaction of nitrate aerosols with residual carbon on the vaporizer surfaces (Pieber et al. Citation2016). With the CV, the measurement of f44 is expected to be less instrument-sensitive than for the SV due to: (i) much faster CO2+ signal decay in the CV—e.g., τ of CO2+ from citric acid is less than 3 s in the CV vs ∼11 s in the SV ( and ). In typical operation, the beam-blocking is switched every ∼4–5 s. Thus, the differential CO2+ signal in the CV is always calculated based on subtracting a lower background than in the SV, leading to less uncertainty. As demonstrated in Figure S13, in MS mode of 5 s beam open vs 5 s beam closed, the CO2+ signal in the CV could be 2 times higher than that in the SV if we only consider the differences result from decay lifetime. The slow decay of CO2+ in the SV is probably due to a large fraction of signal originating from slow vaporization of bounced particles residing on surrounding ion cage surfaces. (ii) A much lower interference (CO2+ per NO3 mass ratio <0.4%) is observed in the CV than the SV (1.5%) when sampling pure NH4NO3 particles (Hu et al. Citation2017b). This should result in more robust CO2+ measurements with the CV when NO3/OA is high (e.g., >1).
m/z 43 signal, typically dominated by C3H7+ and C2H3O+, is another prominent fragment in OA spectra detected in AMS. Ng et al. (Citation2010) showed that the PMF-resolved OA components cluster in a well-defined triangular region in the scatter plot of f44 vs f43, which is widely used to describe the evolution of different types of OA during aging. The relationship of f43 vs f44 in the CV is explored here. For ambient studies (i.e., SOAS, Billerica, and KORUS-AQ), the f43 vs f44 data for the SV cluster in the middle of defined triangle area (); whereas, for the CV ambient data appear in the top of the triangle with lower f43 and higher f44 (). Similar behavior was found for chamber SOA and pure standard OA detected in the CV, albeit with greater variability as expected due to the simpler compositions. A potential new CV “triangle” area is defined based on the measured ambient datasets in .
We also explored the variation of other ions for ambient OA in the CV vs SV. Taking the SOAS study as an example, shows that higher regression slopes (>1) are more frequently obtained at m/z < 50 and lower slopes (<1) occur for majority of m/z > 50, consistent with the average spectra comparison shown in . The degree of signal reduction varies for different m/z—e.g., for the marker of IEPOX-SOA, m/z 82 (C5H6O+, Hu et al. Citation2015), a regression slope of ∼0.7 for f82 between the CV and SV is found, with R∼0.9, suggesting the robustness of that tracer ion for the CV. Good correlation between f82 measured in the SV with other IEPOX-SOA related ions (originally identified for the SV, e.g., m/z 53, 70, Hu et al. Citation2015) measured in the CV is also found (Figure S15). Meanwhile, for another possible biogenic indicator in forest areas, m/z 91, thought to be associated with SOA formed through monoterpene oxidation (e.g., monoterpene + NO3 or O3) (Lee et al. Citation2016), an R∼0.8 with a regression slope of 0.5 is obtained, suggesting that the tracer information is conserved. For the commonly-used biomass burning tracer, f60 (Alfarra et al. Citation2007; Cubison et al. Citation2011), a factor of 5 decrease in the CV compared to the SV was observed. Different reductions of m/z were likely caused by the different vaporized neutral and tracer ion structure, leading to different degrees of additional thermal decomposition and/or ion fragmentation.
Figure 6. (a) Regression slopes and correlation coefficients (R) between time series of different ions between CV and SV during the SOAS study. Scatter plots of several tracer ions (b) f43, (c) f60, (d) f82 and (e) f91 between the CV and SV during the SOAS study. All the regressions of scatter plots are determined using orthogonal distance regression with fits forced through zero intercept and in all figures shown below. The vertical black dashed line is the AMS background f60 value (0.3%), calculated based on various non-BB influenced OA plumes in the field studies (Cubison et al. Citation2011). The inset in (c) shows a time series of f60 from the CV and SV for a short period with significant BB impacts. The grey dots are the entire dataset of f60 with a correlation coefficient R of 0.2. The inset of (d) is the scatter plots between f44 vs f82. The black dashed line is the AMS background value of f82 ( = −0.008 × f44 + 0.005), reported in Hu et al. (Citation2015). The grey dashed line is the eye-guided boundary line for IEPOX-SOA influenced triangle areas determined for the SV (Hu et al. Citation2015).
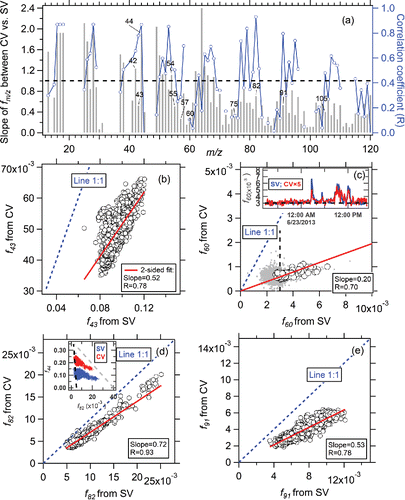
Overall, several important fm/z ( = i.e., the ratio of the organic signal at a given m/z to the total OA signal) show good correlations between the CV and SV, with R > 0.6. Some low correlation coefficients (R < 0.4) are due to the low signal-to-noise level of those ions in the CV, e.g., a correlation coefficient of 0.2 is observed for f60 between the CV and SV when the entire dataset for the SOAS study is considered. However, close covariance of f60 is observed at high values of f60 (during actual BB impact) with R = 0.7, as shown in the inset of . The lowest values of f43, f60, and f91 are much lower in the CV than those in the SV (), suggesting that some interferences on key tracer ions may be reduced in the CV.
We have proposed several new potential biomass burning tracers when BBOA is detected by the CV (e.g., m/z 26: C2H2+; m/z 42: C2H2O+; m/z 56: C3H4O+; m/z 68: C4H4O+; m/z 96: C5H4O+) based on the observed correlation between f60 and BBOA in the SV versus all the fmz measure in the CV and also the measured spectra from biomass burning tracer levoglucosan and sucrose particles in the CV. The details can be found in Section 2 of the supporting information.
In summary, although reduced ion intensities at higher m/z are observed with the CV, it appears that the information for several key tracer ions may be retained.
4. Conclusions
In this study, we have examined the fragmentation pattern of OA in the CV-AMS compared to SV-AMS using multiple pure OA standard compounds, chamber-generated SOA, and ambient OA. A public, web-based spectral database for CV-AMS is newly established. The link of this webpage: http://cires1.colorado.edu/jimenez-group/AMSsd_CV/. Similar to inorganic sulfate and nitrate, fragmentation patterns of OA in the CV also show evidence of additional thermal decomposition, with a shift in the ion distributions to smaller ions. We hypothesize that longer residence times inside the CV will increase internal energy of gas vapors leading to more decomposition. The shift to smaller ions occurs more for the oxygen-containing ions than reduced hydrocarbon ions, due to thermal defunctionalization reactions. We found artificial CO+ production, when sampling long-chain alkane/alkene (e.g., squalene, C30H50; octacosane, C28H58) or similar reduced standards (e.g., oleic acid, C18H36O2) in the CV. Enhanced H2O+ was observed in almost all the OA spectra examined. The extra CO+ (and H2O+ to a lesser extent) is probably caused by the chemical reaction of sampled OA with previously absorbed lattice oxygen on a Mo oxide layer on the vaporizer surface. It was shown that the carbon in the CO+ is derived from the aerosol species impacting the vaporizer. This artificial CO+ ion production was not observed when CV-AMSs sample ambient OA from field studies; thus, elemental ratios and PMF analysis for those aerosols should not be impacted (except possibly for HOA, which was not abundant on the studies analyzed here). The lack of artifact CO+ for more oxidized compounds may be due to the oxidized species readily undergoing prompt thermal decomposition on the vaporizer, while alkanes and alkenes do not promptly decompose, and thus can take part in slower oxidation reactions on the vaporizer surface. Investigation of CVs built of other materials is recommended for the future studies.
Strong correlations for tracer fm/z are observed between the CV and SV during the SOAS ambient study, suggesting most of the characteristic ion tracers identified in the SV can still be used for CV measurements. A potential new f44- f43 triangle plot for CV is also proposed. Reduced signals for biomass burning tracers f60 and f73 (∼ a factor of 5) are found in the CV due to strong thermal decomposition effects. While these ions continue to be useful as BB tracers, their application may be limited at low signal or BB contribution levels. m/z 26 (C2H2+), 42 (C2H2O+), 56 (C3H4O+), 68 (C4H4O+), 96 (C5H4O+), could be potentially new biomass burning tracers for CV-AMS, and should be further investigated with ambient or chamber studies which are (and are not) strongly influenced by biomass burning emissions.
UAST_1454584_Supplementary_file.zip
Download Zip (2.2 MB)Acknowledgments
We sincerely thank Uwe Karst, Tim Elseberg, and Teledyne CETAC for the use of the laser ablation setup for our experiment. We thank the DC-8 crew for support of the KORUS-AQ study.
Additional information
Funding
References
- Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L. (2008). O/C and OM/OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High-Resolution Time-of-Flight Aerosol Mass Spectrometry. Environ. Sci. Technol., 42:4478–4485. doi:10.1021/Es703009q.
- Aimanant, S. and Ziemann, P. J. (2013). Chemical Mechanisms of Aging of Aerosol Formed from the Reaction of n-Pentadecane with OH Radicals in the Presence of NOx. Aerosol. Sci. Tech., 47:979–990. doi:10.1080/02786826.2013.804621.
- Alfarra, M. R., Coe, H., Allan, J. D., Bower, K. N., Boudries, H., Canagaratna, M. R., Jimenez, J. L., Jayne, J. T., Garforth, A. A., Li, S. M., and Worsnop, D. R. (2004). Characterization of Urban and Rural Organic Particulate in the Lower Fraser Valley Using two Aerodyne Aerosol Mass Spectrometers. Atmos. Environ., 38:5745–5758. doi:10.1016/j.atmosenv.2004.01.054.
- Alfarra, M. R., Prevot, A. S. H., Szidat, S., Sandradewi, J., Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U. (2007). Identification of the Mass Spectral Signature of Organic Aerosols from Wood Burning Emissions. Environ. Sci. Technol., 41:5770–5777. doi:10.1021/Es062289b.
- Allan, J. D., Alfarra, M. R., Bower, K. N., Williams, P. I., Gallagher, M. W., Jimenez, J. L., McDonald, A. G., Nemitz, E., Canagaratna, M. R., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003a). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer – 2. Measurements of Fine Particulate Chemical Composition in two U.K. Cities. J. Geophys. Res-Atmos., 108(D3):4091. doi:10.1029/2002jd002359.
- Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R. (2003b). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer: 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res-Atmos., 108. doi:10.1029/2003jd001607.
- Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R. (2007). Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer. Mass. Spectrom. Rev., 26:185–222. doi:10.1002/Mas.20115.
- Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R. (2015). Elemental Ratio Measurements of Organic Compounds Using Aerosol Mass Spectrometry: Characterization, Improved Calibration, and Implications, Atmos. Chem. Phys., 15:253–272. doi:10.5194/acp-15-253-2015.
- Carlton, A. M., de Gouw, J., Jimenez, J. L., Ambrose, J. L., Brown, S., Baker, K. R., Brock, C. A., Cohen, R. C., Edgerton, S., Farkas, C., Farmer, D., Goldstein, A. H., Gratz, L., Guenther, A., Hunt, S., Jaeglé, L., Jaffe, D. A., Mak, J., McClure, C., Nenes, A., Nguyen, T. K. V., Pierce, J. R., Selin, N., Shah, V., Shaw, S., Shepson, P. B., Song, S., Stutz, J., Surratt, J., Turpin, B. J., Warneke, C., Washenfelder, R. A., Wennberg, P. O., and Zhou, X. (2018). The Southeast Atmosphere Studies (Sas): Coordinated Investigation and Discovery to Answer Critical Questions About Fundamental Atmospheric Processes. Bull. Am. Met. Soc., In press
- Crenn, V., Sciare, J., Croteau, P. L., Verlhac, S., Fröhlich, R., Belis, C. A., Aas, W., Äijälä, M., Alastuey, A., Artiñano, B., Baisnée, D., Bonnaire, N., Bressi, M., Canagaratna, M., Canonaco, F., Carbone, C., Cavalli, F., Coz, E., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Lunder, C., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petit, J. E., Petralia, E., Poulain, L., Priestman, M., Riffault, V., Ripoll, A., Sarda-Estève, R., Slowik, J. G., Setyan, A., Wiedensohler, A., Baltensperger, U., Prévôt, A. S. H., Jayne, J. T., and Favez, O. (2015). ACTRIS ACSM Intercomparison – Part 1: Reproducibility of Concentration and Fragment Results from 13 Individual Quadrupole Aerosol Chemical Speciation Monitors (Q-Acsm) and Consistency with Co-Located Instruments. Atmos. Meas. Tech., 8:5063–5087. doi:10.5194/amt-8-5063-2015.
- Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L. (2011). Effects of Aging on Organic Aerosol from Open Biomass Burning Smoke in Aircraft and Laboratory Studies. Atmos. Chem. Phys., 11:12049–12064. doi:10.5194/acp-11-12049-2011.
- Davidson, C. I., Phalen, R. F., and Solomon, P. A. (2005). Airborne Particulate Matter and Human Health: A Review. Aerosol. Sci. Tech., 39:737–749. doi:10.1080/02786820500191348.
- DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal. Chem., 78:8281–8289. doi:10.1021/Ac061249n.
- Drewnick, F., Jayne, J. T., Canagaratna, M., Worsnop, D. R., and Demerjian, K. L. (2004). Measurement of Ambient Aerosol Composition During the Pmtacs-Ny 2001 Using an Aerosol Mass Spectrometer. Part II: Chemically Speciated Mass Distributions. Aerosol. Sci. Tech., 38:104–117. doi:10.1080/02786820390229534.
- Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R. (2005). A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS) – Instrument Description and First Field deployment. Aerosol. Sci. Tech., 39:637–658. doi:10.1080/02786820500182040.
- Drewnick, F., Diesch, J. M., Faber, P., and Borrmann, S. (2015). Aerosol Mass Spectrometry: Particle–Vaporizer Interactions and their Consequences for the Measurements. Atmos. Meas. Tech., 8:3811–3830. doi:10.5194/amt-8-3811-2015.
- Duplissy, J., DeCarlo, P. F., Dommen, J., Alfarra, M. R., Metzger, A., Barmpadimos, I., Prevot, A. S. H., Weingartner, E., Tritscher, T., Gysel, M., Aiken, A. C., Jimenez, J. L., Canagaratna, M. R., Worsnop, D. R., Collins, D. R., Tomlinson, J., and Baltensperger, U. (2011). Relating Hygroscopicity and Composition of Organic Aerosol Particulate Matter. Atmos. Chem. Phys., 11:1155–1165. doi:10.5194/acp-11-1155-2011.
- Fröhlich, R., Crenn, V., Setyan, A., Belis, C. A., Canonaco, F., Favez, O., Riffault, V., Slowik, J. G., Aas, W., Aijälä, M., Alastuey, A., Artiñano, B., Bonnaire, N., Bozzetti, C., Bressi, M., Carbone, C., Coz, E., Croteau, P. L., Cubison, M. J., Esser-Gietl, J. K., Green, D. C., Gros, V., Heikkinen, L., Herrmann, H., Jayne, J. T., Lunder, C. R., Minguillón, M. C., Močnik, G., O'Dowd, C. D., Ovadnevaite, J., Petralia, E., Poulain, L., Priestman, M., Ripoll, A., Sarda-Estève, R., Wiedensohler, A., Baltensperger, U., Sciare, J., and Prévôt, A. S. H. (2015). ACTRIS ACSM Intercomparison – Part 2: Intercomparison of ME-2 Organic Source Apportionment Results from 15 Individual, Co-Located Aerosol Mass Spectrometers. Atmos. Meas. Tech., 8:2555–2576. doi:10.5194/amt-8-2555-2015.
- Gulbransen, E. A., Andrew, K. F., and Brassart, F. A. (1963). Oxidation of Molybdenum 550°C to 1700°C. J. Electrochem. Soc., 110:952–959. doi:10.1149/1.2425918.
- Gunther, D. and Heinrich, A. C. (1999). Enhanced Sensitivity in Laser Ablation-Icp Mass Spectrometry Using Helium-Argon Mixtures as Aerosol Carrier. Journal of Analytical Atomic Spectrometry., 14:1363–1368. doi:10.1039/a901648a.
- Hu, W., Campuzano-Jost, P., Day, D. A., Croteau, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L. (2017a). Evaluation of the New Capture Vaporizer for Aerosol Mass Spectrometers (Ams) Through Field Studies of Inorganic Species. Aerosol. Sci. Tech., 51:735–754. doi:10.1080/02786826.2017.1296104.
- Hu, W., Campuzano-Jost, P., Day, D. A., Croteau, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L. (2017b). Evaluation of the New Capture Vapourizer for Aerosol Mass Spectrometers (Ams) through Laboratory Studies of Inorganic Species. Atmos. Meas. Tech., 10:2897–2921. doi:10.5194/amt-10-2897-2017.
- Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L. (2015). Characterization of a Real-Time Tracer for Isoprene Epoxydiols-Derived Secondary Organic Aerosol (Iepox-Soa) from Aerosol Mass Spectrometer Measurements. Atmos. Chem. Phys., 15:11807–11833. doi:10.5194/acp-15-11807-2015.
- IPCC. (2017). Intergovernmental Panel on Climate Change (IPCC): Climate Change: The Scientific Basis. Cambridge University Press, UK.
- Jayne, J. T. and Worsnop, D. R. (2016). Particle capture device. Aerodyne Research, Inc. 20150040689 A1
- Jimenez, J. L., Bahreini, R., Cocker, D. R., Zhuang, H., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., O'Dowd, C. D., and Hoffmann, T. (2003). New Particle Formation from Photooxidation of Diiodomethane (CH2I2). J. Geophys. Res-Atmos., 108:4318. doi:10.1029/2003jd004249.
- Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R. (2009). Evolution of Organic Aerosols in the Atmosphere. Science., 326:1525–1529. doi:10.1126/science.1180353.
- Jimenez, J. L., Canagaratna, M. R., Drewnick, F., Allan, J. D., Alfarra, M. R., Middlebrook, A. M., Slowik, J. G., Zhang, Q., Coe, H., Jayne, J. T., and Worsnop, D. R. (2016). Comment on “The Effects of Molecular Weight and Thermal Decomposition on the Sensitivity of a Thermal Desorption Aerosol Mass Spectrometer”. Aerosol. Sci. Tech., 50:i–xv. doi:10.1080/02786826.2016.1205728.
- Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J. (2005). Organic Aerosol and Global Climate Modelling: A Review. Atmos. Chem. Phys., 5:1053–1123.
- Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E. (2015). Sources, Seasonality, and Trends of Southeast us Aerosol: An Integrated Analysis of Surface, Aircraft, and Satellite Observations with the Geos-Chem Chemical Transport Model. Atmos. Chem. Phys., 15:10411–10433. doi:10.5194/acp-15-10411-2015.
- Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., St. Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R. (2015). Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation. Environ. Sci. Technol., 49:10330–10339. doi:10.1021/acs.est.5b02031.
- Krechmer, J. E., Day, D. A., Ziemann, P. J., and Jimenez, J. L. (2017). Direct Measurements of Gas/Particle Partitioning and Mass Accommodation Coefficients in Environmental Chambers, Environ. Sci. Technol., 51, 11867–11875. doi:10.1021/acs.est.7b02144.
- Lee, A. K. Y., Abbatt, J. P. D., Leaitch, W. R., Li, S. M., Sjostedt, S. J., Wentzell, J. J. B., Liggio, J., and Macdonald, A. M. (2016). Substantial Secondary Organic Aerosol Formation in a Coniferous Forest: Observations of Both day- and Nighttime Chemistry. Atmos. Chem. Phys., 16:6721–6733. doi:10.5194/acp-16-6721-2016.
- Liao, J., Brock, C. A., Murphy, D. M., Sueper, D. T., Welti, A., and Middlebrook, A. M. (2017). Single-Particle Measurements Of Bouncing Particles And In Situ Collection Efficiency from an Airborne Aerosol Mass Spectrometer (AMS) With Light-Scattering Detection. Atmos. Meas. Tech., 10:3801–3820. doi:10.5194/amt-10-3801-2017.
- Matthew, B. M., Middlebrook, A. M., and Onasch, T. B. (2008). Collection Efficiencies in an Aerodyne Aerosol Mass Spectrometer as a Function of Particle Phase for Laboratory Generated Aerosols. Aerosol. Sci. Tech., 42:884–898. doi:10.1080/02786820802356797.
- Mauderly, J. L. and Chow, J. C. (2008). Health Effects of Organic Aerosols. Inhal Toxicol., 20:257–288. doi:10.1080/08958370701866008.
- Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R. (2012). Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data. Aerosol. Sci. Tech., 46:258–271. doi:10.1080/02786826.2011.620041.
- Moldoveanu, S. (2009). Pyrolysis of Organic Molecules: Applications to Health and Environmental Issues. Elsevier Science.
- Murphy, D. M., Cziczo, D. J., Froyd, K. D., Hudson, P. K., Matthew, B. M., Middlebrook, A. M., Peltier, R. E., Sullivan, A., Thomson, D. S., and Weber, R. J. (2006). Single-Particle Mass Spectrometry of Tropospheric Aerosol Particles. Journal of Geophysical Research: Atmospheres., 111, n/a-n/a. doi:10.1029/2006jd007340.
- Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prevot, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R. (2010). Organic Aerosol Components Observed in Northern Hemispheric Datasets from Aerosol Mass Spectrometry. Atmos Chem Phys., 10:4625–4641. doi:10.5194/acp-10-4625-2010.
- Ng, N. L., Jayne, J. T., Herndon, S. C., Trimborn, A., Canagaratna, M. R., Croteau, P. L., Onasch, T. B., Sueper, D., Worsnop, D. R., Zhang, Q., and Sun, Y. L. (2011). An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol. Aerosol Sci Tech., 45:780–794. doi:10.1080/02786826.2011.560211.
- Ozkan, U. S., Driscoll, S. A., Zhang, L., and Ault, K. L. (1990a). Investigation of oxygen insertion mechanism in selective oxidation reactions over MnMoO4/MoO3 catalysts through isotopic labeling and chemisorption studies. J Catal., 124:183–193. https://doi.org/10.1016/0021-9517(90)90113-X.
- Ozkan, U. S., Smith, M. R., and Driscoll, S. A. (1990b). Effect of O2 Concentration on Selective and Complete Oxidation of 1,3-Butadiene, Furan, and Maleic Anhydride Over MnMoO4/MoO3 Catalysts. J Catal., 123:173–180. https://doi.org/10.1016/0021-9517(90)90166-H.
- Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Fröhlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jiménez, J. L., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H. (2016). Inorganic Salt Interference on CO2+ in Aerodyne AMS and ACSM Organic Aerosol Composition Studies. Environ. Sci. Technol., 50:10494–10503. doi:10.1021/acs.est.6b01035.
- Poschl, U. (2005). Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects. Angew. Chem. Int. Edit., 44:7520–7540. doi:10.1002/anie.200501122.
- Reifschneider, O., Wentker, K. S., Strobel, K., Schmidt, R., Masthoff, M., Sperling, M., Faber, C., and Karst, U. (2015). Elemental Bioimaging of Thulium in Mouse Tissues by Laser Ablation-ICPMS as a Complementary Method to Heteronuclear Proton Magnetic Resonance Imaging for Cell Tracking Experiments. Anal. Chem., 87:4225–4230. doi:10.1021/ac504363q.
- Robinson, E. S., Onasch, T. B., Worsnop, D., and Donahue, N. M. (2017). Collection Efficiency of α-pinene Secondary Organic Aerosol Particles Explored Via Light-Scattering Single-Particle Aerosol Mass Spectrometry. Atmos. Meas. Tech., 10:1139–1154. doi:10.5194/amt-10-1139-2017.
- Salcedo, D., Onasch, T. B., Aiken, A. C., Williams, L. R., de Foy, B., Cubison, M. J., Worsnop, D. R., Molina, L. T., and Jimenez, J. L. (2010). Determination of Particulate Lead Using Aerosol Mass Spectrometry: MILAGRO/MCMA-2006 Observations, Atmos. Chem. Phys., 10:5371–5389. doi:10.5194/acp-10-5371-2010.
- Tobias, H. J., Kooiman, P. M., Docherty, K. S., and Ziemann, P. J. (2000). Real-Time Chemical Analysis of Organic Aerosols Using a Thermal Desorption Particle Beam Mass Spectrometer. Aerosol. Sci. Tech., 33:170–190. doi:10.1080/027868200410912.
- Voisin, D., Smith, J. N., Sakurai, H., McMurry, P. H., and Eisele, F. L. (2003). Thermal Desorption Chemical Ionization Mass Spectrometer for Ultrafine Particle Chemical Composition. Aerosol. Sci. Tech., 37:471–475. doi:10.1080/02786820300959.
- Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L. (2014). Effects of Anthropogenic Emissions on Aerosol Formation from Isoprene and Monoterpenes in the Southeastern United States. Proceedings of the National Academy of Sciences., 112:37–42. doi:10.1073/pnas.1417609112.
- Xu, W., Croteau, P., Williams, L., Canagaratna, M., Onasch, T., Cross, E., Zhang, X., Robinson, W., Worsnop, D., and Jayne, J. (2017). Laboratory Characterization of an Aerosol Chemical Speciation Monitor with PM2.5 Measurement Capability. Aerosol. Sci. Tech., 51:69–83. doi:10.1080/02786826.2016.1241859.
- Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R. (2007). Ubiquity and Dominance of Oxygenated Species in Organic Aerosols in Anthropogenically-Influenced Northern Hemisphere Midlatitudes. Geophys. Res. Lett., 34:L13801. doi:10.1029/2007gl029979.