To the Editor
Extraskeletal Ewing sarcoma (EES) is a rare soft tissue tumour that is morphologically indistinguishable from the more common Ewing sarcoma of bone. It must be differentiated from other small, blue round cell tumours, including primitive neuroectodermal tumour and neuroblastoma. The most frequent sites of occurrence are the chest wall, lower extremities, and paravertebral region. Less frequently, the tumour occurs in the pelvis and hip region, the retroperitoneum and the upper extremities Citation[1], Citation[2]. The age at the time of diagnosis, unlike its osseous counterpart, has a wide range, from infancy to the elderly, and has a slight predominance in male patient Citation[2], Citation[3].
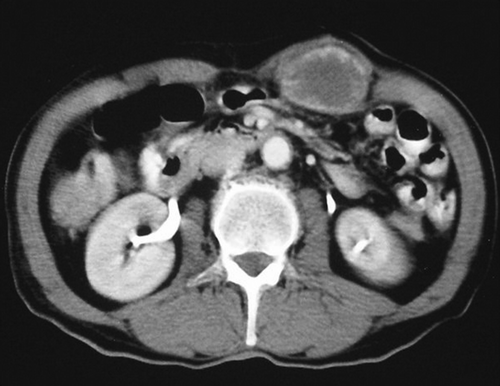
A 65-year old man presented with a painless mass in the abdominal wall. There was no history of weight loss, trauma or infection for the last year. Physical examination revealed an irregular mass approximately 5 cm in diameter in the left upper quadrant of the abdominal wall. Other laboratory results were normal. Contrast enhanced axial CT scan showed a 5 cm in diameter hypodense mass with peripheral contrast enhancement in the left rectus muscle ().
Figure 1. Post contrast axial CT scan shows hypodense with peripheral contrast enhancement mass in the left rectus muscle.
An ultrasound-guided fine needle aspiration for cytological examination was performed and it revealed diffuse lymphoma. To confirm the diagnosis, open biopsy of the mass was done. Histopathological examination showed a neoplasm with focal areas of fibrosis with a vague trabecular pattern, existing necrosis and fairly uniform, round, blue, medium-sized cells in a diffuse pattern (). The vacuolated cytoplasm of the tumour cells showed a high content of glycogen demonstrable by Periodic acid-Schiff (PAS). The tumour was negative for epithelial membrane antigen, high and low molecular weight cytokeratin, CD20, CD68, leukocyte common antigen, desmin, muscle specific actin. Immunohistochemical evidence of CD99 in Ewing sarcoma, were strongly positive, confirming extraosseous Ewing sarcoma. A99m technetium methylene diphosphonate whole body bones scan showed normal skeletal uptake of tracer and no evidence of bone involvement. The mass and entire left rectus muscle bellies were resected via a left paramedian incision. Becoming defect was covered by composix mesh (). The postoperative course was uneventful, and the patient was discharged from the hospital at the tenth postoperative day. One month after the discharge chemotherapy was carried out including vincristine, doxorubicine, cyclophosphamide, and etoposide for six cycles, and the patient has had no recurrence of the disease during at the one year follow-up.
Figure 2. Ewing sarcoma, comprising a highly cellular sheet of relatively featureless small cells, with round, dark nuclei and inconspicuous cytoplasm (H×E 20×10).

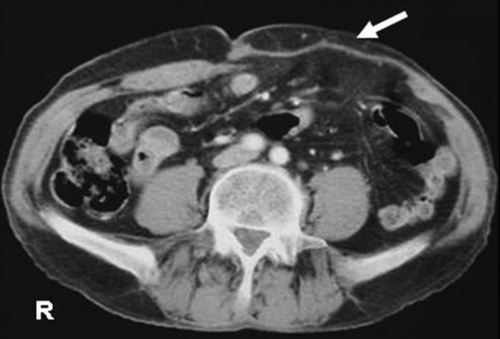
Figure 3. Postoperative axial CT scan shows composix mesh (arrow) in the left abdominal wall.
Ewing sarcoma commonly arises from bone, but rarely may have an extraskeletal origin. EES is a rare soft tissue tumour that is morphologically indistinguishable from the more common Ewing sarcoma of bone. It can be confused with other small, round, blue cell tumours, including embryonal rhabdomyosarcoma, lymphoma, and neuroblastoma Citation[2], Citation[4]. The most frequent sites of occurrence are the chest wall, lower extremities, and paravertebral region. Less frequently, the tumour occurs in the pelvis and hip region, the retroperitoneum, and the upper extremities Citation[2]. The age at the time of diagnosis, unlike its osseous counterpart, has a wide range, from infancy to the elderly, and has a slight predominance in male patients Citation[2], Citation[3]. We did a literature search using key search term “Ewing sarcoma”, “primitive neuroectodermal tumour”, and “abdominal wall” and there were no cases with Ewing sarcoma of the abdominal wall in the Medline.
This tumour shares histologic, immunohistochemical, and molecular findings with ES of bone Citation[5]. The classical histologic description of Extraskeletal Ewing sarcoma is of a tumour composed of small, uniform, round or oval cells in solid sheets divided by fibrous strands. Its cytoplasm is scanty, pale staining, and vacuolated because of the presence of glycogen, and the nuclei are round with “salt and pepper” chromatin and small nucleoli. A sensitive and relatively specific antigen, CD99/MIC2, and a characteristic chromosomal translocation, t (11; 22), have been identified in skeletal ES and EES Citation[5].
Initially, extraosseous Ewing sarcoma classified as a soft tissue sarcoma and treated like rhabdomyosarcoma on IRS-I to III (Intergroup Rhabdomyosarcoma Study) Citation[6], but more recently, they have been classified and treated as osseous Ewing sarcomas Citation[6]. The chemotherapy of this protocol has been similar. One substantial difference is the surgical approach; IRS trials incorporate initial surgical resection, while Ewing sarcoma trials typically use neoadjuvant chemotherapy with delayed surgery Citation[6]. Rud et al. Citation[3] found that aggressive surgical treatment was significant. Raney et al. Citation[4] also reported an increased survival with macroscopically complete tumour removal prior to the initiation of chemotherapy.
The results of a study by Ahmad et al. Citation[3] demonstrate that EES is a curable disease and has a best prognosis in patients aged < 16 years who are treated with adequate resection in conjunction with chemotherapy. CT and MRI are used for determinate the site of origin of the tumour and metastases, which are also evaluate the rate of resectability. When resectable, the surgical excision must be wide in accordance to the classic oncological principles. The defect may be repaired by direct approximation of abdominal wall edges or this may require prosthetic repair of wall. In our patient after the mass was removed, the defect was repaired with a composix mesh.
Metastases are present at diagnosis in 9 to 20% of patients Citation[6]. The lungs are the most common site of metastases and treatment for metastatic disease is early aggressive combination chemotherapy rather than single agent monotherapy Citation[5]. As these tumours are also radiosensitive; tumours that are not appropriate to surgical resection or have positive margins, are treated with radiation Citation[6].
The 5-year disease-free survival is 24 to 80% for localized disease, with smaller resectable lesions Citation[6]. Raney et al. Citation[3] reported a 10-year survival rate between 61 and 77% utilizing multiagent chemotherapy, which was similar to overall 5-year survival rate of 61% found in the current studies. Our patient was treated successfully with surgery and multiple-agent, chemotherapy, and no recurrence was observed in the one year follow up.
EES is a rare tumour and sensitive to multimodality treatment. All patients should be treated with multiagent chemotherapy and aggressive surgical treatment. EES is a curable disease even when metastatic disease is present at the time of diagnosis Citation[3].
References
- Rud NP, Reiman H, Pritchard DT, Frassica FJ, Smithson WA. Extraosseous Ewing's sarcoma: A study of cases. Cancer 1989; 64: 1548–53
- Cheung CC, Kandel RA, Bell RS, Mathews RE, Ghazarian MD. Extraskeletal Ewing sarcoma in a 77-year-old woman. Arc Pathol Lab Med 2001; 125: 1358–60
- Ahmad R, Brian R, Davis M, Bruce T. Extraskeletal Ewing's sarcoma. Cancer 1999; 85: 725–31
- Toh KL, Tan PH, Cheng WS. Primary Extraskeletal Ewing's sarcoma of the external genitalia. J Urol 1999; 62: 159–60
- Guiter GE, Gamboni MM, Zakowski MF. The cytology of extraskeletal Ewing sarcoma. Cancer 1999; 87: 141–8
- Gaona-Luviano P, Unda-Franco E, Gonzalez-Jara L, Romero P, Medina-Franco H. Primitive neuroectodermal tumour of the vagina. Gynecol Oncol 2003; 91: 456–8