Extensive research focusing on viral vectors has allowed for significant advances in the field of gene therapy. Oncolytic adenoviruses may in the near future become important therapeutic agents in the management of organ-confined prostate cancer. An adenovirus, whose replication is controlled by a prostate-specific regulatory sequence, specifically replicates in and lyses cells of prostate epithelial origin, including prostate adenocarcinoma cells. Such a virus offers two appealing properties: it specifically kills normal and neoplastic prostate cells and its replication leads to controlled virus spread and thereby amplification of the therapeutic effect. Intraprostatic injections of oncolytic adenoviruses have shown good safety profiles and promising potential when combined with external radiation therapy. Current development of oncolytic viruses focuses on how to further improve their efficacy by including therapeutic genes or genes that can modulate the immune response mounted against them. Recent discoveries in tumor immunology have renewed hopes that immunotherapy may play a role in the management of advanced prostate cancer. One interesting approach to overcome the immunological tolerance that appears to be firmly established in many advanced tumors is ex vivo expansion and adoptive transfer of prostate antigen-specific cytolytic T lymphocytes. In parallel, extensive research on cancer vaccine development based on dendritic cells, tumor antigens, cytokines, and novel immune adjuvants is currently ongoing. It will be essential to include immunotherapy protocols early on in the course of recurrent prostate cancer in order to extend survival and improve the quality of life.
Prostate cancer
Prostate cancer is a leading cause of illness and death among men in Western countries. Current statistics from the American Cancer Society estimate that 220 900 men will be diagnosed with prostate cancer and that 28 900 will die from this disease in the USA in 2003 Citation[1]. The current standard therapies employed for organ-confined prostate cancer include radical prostatectomy, external beam irradiation, and brachytherapy, under some circumstances incorporating neoadjuvant or adjuvant hormonal therapy Citation[2], Citation[3]. While these therapies are relatively effective in the short term, a significant proportion (30–40%) of patients who initially present with localized disease ultimately relapse Citation[4]. Moreover, these therapies are often associated with significant side effects due to injury to adjacent tissues. For metastatic prostate cancer, the main therapy is androgen ablation. While this provides cytoreduction and palliation, progression to hormone-refractory disease typically occurs within the order of 14–20 months. A large number of clinical studies have been reported in the field of chemotherapy for advanced androgen-independent prostate cancer. Only recently have two trials revealed that chemotherapy marginally improves the overall survival of patients with hormone-refractory disease Citation[5], Citation[6]. Thus, there is a need for improved techniques to control localized prostate cancer and treat advanced hormone-refractory prostate cancer, which can complement the standard techniques.
The tendency for increased prostate cancer screening, typically with digital rectal examination and measurement of serum prostate-specific antigen (PSA), means that prostate cancer can be diagnosed at an earlier stage than was previously possible Citation[7]. In the USA the clinical picture of prostate cancer is very different today from what it was 10–15 years ago, with a higher proportion of men with organ-confined disease and negative surgical margins Citation[8]. In Sweden, where systematic PSA screening has not yet been initiated and where prostate cancer is still most commonly diagnosed during the work-up of symptomatic patients Citation[9], such a clinical shift has not yet been observed. However, recent studies clearly indicate that early detection and early removal of the cancerous cells would be advantageous for prostate cancer patients Citation[10]. Therefore, PSA testing will probably increase in Sweden, reducing the death toll from prostate cancer it is hoped, though at the same time it will cause a dramatic increase in the demand for organ-confined prostate cancer treatments.
Since the prostate gland is an organ not essential for life, proteins that are specifically expressed by neoplastic (and normal) prostate cells constitute suitable targets for prostate cancer immunotherapy. In addition, gene promoters of such prostate-specific proteins are transcriptionally active only in normal and neoplastic prostate cells and may therefore be included in viral vectors to restrictively control expression of therapeutic molecules or control replication of oncolytic viruses. Prostate cancer is particularly appropriate for gene therapy and immunotherapy, since a large number of genes and proteins with specific or preferential expression in prostate and prostate cancer cells have been identified, including prostate specific antigen (PSA) Citation[11], kallikrein 2 Citation[12], prostatic acid phosphatase (PAP) Citation[13], prostate-specific membrane antigen (PSMA) Citation[14], prostate-specific transglutaminase Citation[15], homeobox NKX3.1 Citation[16], prostate stem cell antigen (PSCA) Citation[17], PAGE Citation[18], TARP Citation[19], Citation[20], DD3 Citation[21], STEAP Citation[22], prostase/kallikrein 4 Citation[23], PDEF Citation[24], hPSE Citation[25], PCGEM1 Citation[26], PART-1 Citation[27], PSGR Citation[28], PMEPA1 Citation[29], prostein Citation[30], GDEP Citation[31], Trp-p8 Citation[32], PSDR1 Citation[33], Cten Citation[34], PATE Citation[35], POTE Citation[36], STAMP-1 Citation[37], AIbZIP Citation[38], HPG-1 Citation[39], NGEP Citation[40], and PrLZ Citation[41]. Furthermore, many of the promoters controlling expression of these genes have been cloned and characterized.
Gene therapy
Gene therapy is defined as the introduction of a gene coding for a molecule that can cure or slow down the progression of a disease. It may be intended for gene correction, i.e., to replace a missing or non-functional gene, or to carry out gene-mediated destruction of diseased cells. Gene correction is an attractive approach for the treatment of monogenetic inherited diseases. However, in the case of cancer, where there are several genetic alterations, it is far more complicated to revert the malignant phenotype of a tumor cell through gene corrections. Instead, an attractive approach is to deliver a “therapeutic” gene which encodes a molecule that directly or indirectly kills tumor cells. Both viral and non-viral vectors have been developed for gene transfer to tumor cells. Gene transfer vectors have been extensively reviewed elsewhere Citation[42–44]. This review will focus on the use of adenoviral vectors for gene delivery, and in particular adenoviral vectors for gene delivery to prostate cancer cells.
A general review on clinical gene therapy trials worldwide 1989–2004 was recently published Citation[45] and several reviews focusing on cancer gene therapy trials have been published Citation[46–49]. Furthermore, information about clinical trial protocols for prostate cancer gene therapy can be found at the NIH webpage (http://www.gemcris.od.nih.gov/).
Adenoviral vectors
The adenovirus genome is composed of a linear double-stranded DNA molecule of approximately 36 kb. Adenovirus genes are transcribed in a complex temporal manner which can be divided into an early and a late phase, respectively occurring before and after virus DNA replication Citation[50]. Early viral gene expression starts at the E1 locus and includes gene products that modulate cellular metabolism in order to make the infected cell more susceptible to virus replication. The E1A proteins interfere with the processes of cell division by binding to members of the cellular retinoblastoma protein [pRb] family. Blockage of pRb leads to the release of transcription factors of the E2F family that activate transcription of target genes required for entering the S phase of the cell cycle. To circumvent premature apoptotic cell death due to excessive E1A-induced transcriptional activity, the E1B-55kD protein binds to p53 and inhibits its apoptotic activity while the E1B-19kD protein functions as a viral homologue of the cellular anti-apoptotic protein Bcl-2. E1A also plays a central role in activating the adenoviral gene expression cascade. Therefore, viruses lacking E1A are replication defective. The remaining early regions (E2, E3 and E4) encode proteins involved in viral DNA synthesis (E2), modulation of the host immune response and cell lysis (E3), and the regulation of viral gene expression, mRNA transport, DNA replication, and apoptosis (E4). The late gene (L1–L5) products encode viral structural proteins and proteins involved in virion assembly Citation[50].
Adenoviruses can infect both dividing and non-dividing cells in a wide variety of cell types and tissues. This characteristic, together with their relative ease of preparation and purification, has led to their extensive use as gene transfer vectors. The adenovirus genome does not integrate into the host cell genome. Instead it remains episomal in the nucleus, thereby minimizing the risks for recombination and disruption of critical endogenous genes or formation of undesired viral mutants. The first generation of replication-defective adenoviral vectors have the E1 and E3 genes removed, allowing for the introduction of up to 7 kb of foreign DNA. The E1 proteins are then provided in trans by producer cells such as 293, 911, or PerC6. The E3 region is not necessary for replication of adenovirus in vitro and may therefore be abolished. Later generations of replication-defective adenoviral vectors have E2 and/or E4 gene deletions, allowing for the introduction of larger pieces of foreign DNA Citation[50]. Helper-dependent adenoviral vectors that are completely devoid of all viral protein-coding sequences have also been developed Citation[51].
Replication-competent adenoviruses include, at least, the E1A, E2, and E4 regions together with the late genes. They have the ability to infect cells, replicate therein, form progeny viruses that are released through lysis of the infected cell, and spread to neighboring cells. Since the virus itself exerts oncolytic activity, the use of replicating viruses is also referred to as virotherapy. Replication-competent adenoviruses have an advantage over replication-defective adenoviral vectors in the sense that every infected cell serves as a virus-producing cell, thereby augmenting the spread of therapeutic gene expression. Generally speaking, replication-competent viruses raise serious safety concerns about toxicity, risk of mutagenesis, and spread from one patient to other human subjects. However, the type 5 adenovirus meets the safety criteria as it is already prevalent in the human population, is of low pathogenicity, and is a non-integrating virus.
In order to make replication-competent adenoviruses tumor- or tissue-specific, the E1A gene, which controls adenovirus replication by controlling the viral gene expression cascade, can be placed under transcriptional control of tumor- or tissue-specific promoters. These viruses are replication-competent under certain conditions and are referred to as conditionally replication-competent adenoviruses (CRAds). An alternative way to produce tumor CRAds is to develop genetically modified adenoviruses that preferentially replicate in tumor cells with alterations in certain cell cycle pathways, such as the p53 and pRb pathways. Yet another way to restrict oncolytic viruses to certain tumors and tissues is to retarget virus infection either through genetic modification of the viral surface proteins that mediate viral attachment and cell entry, or by using bi-specific linkers that bridge virus surface proteins to specific cell surface receptors (see ).
Figure 1. Strategies for adenovirus retargeting. Restricted adenovirus replication can be achieved through tumor- or tissue-specific promoter-controlled E1A gene expression (a), or through genetic modification of E1A and/or E1B to create virus strains with selective replication in tumor cells with deregulated cell cycle pathways (b). Adenovirus infection can be retargeted from its natural binding receptor to a specific cell surface molecule through genetic modification of the virus fiber or capsid proteins which the virus uses for attachment and cell entry (c) or by using bi-specific linkers that bridge virus surface proteins to specific cell surface receptors (d).
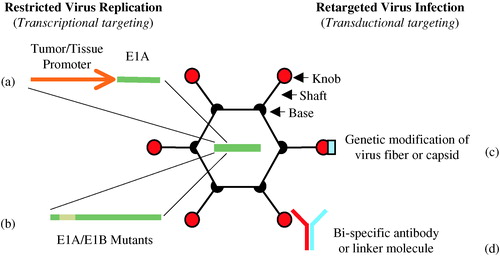
Targeting adenoviral replication to tumor cells using tumor- and tissue-specific promoters
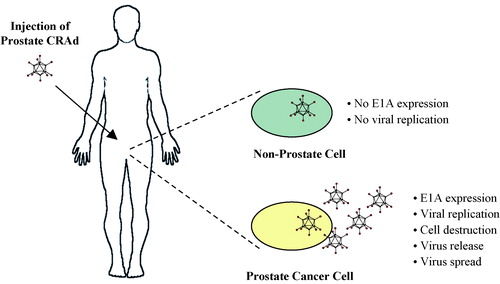
A number of therapeutic approaches relying on prostate-specific transcriptional elements have been envisioned, including therapeutic viruses whose replication is limited to prostate cells and viral vectors with therapeutic genes expressed under the control of prostate-specific regulatory sequences. A prostate CRAd, e.g. an adenovirus with E1A expression under transcriptional control of a prostate-specific regulatory sequence, may be injected intraprostatically to treat localized prostate cancer (see ). The virus will specifically replicate in prostate epithelial cells and prostate cancer cells, leading to lysis of infected cells and viral spread to neighboring cells that are in turn infected and lysed. Alternatively, the prostate CRAd may be injected intravascularly in an effort to treat metastatic prostate cancer.
Figure 2. Oncolytic adenovirus therapy. A conditionally replication-competent adenovirus (CRAd) with E1A gene expression controlled by a prostate-specific promoter specifically replicates in and lyses cells of prostate epithelial origin, including prostate adenocarcinoma cells. The oncolytic prostate CRAd virus may be injected intraprostatically in patients with localized prostate cancer or intravascularly in patients with metastatic prostate cancer.
Henderson and colleagues pioneered the work of prostate CRAds by using promoter and enhancer elements from human kallikrein 2 (hK2), prostate-specific antigen (PSA) and rat probasin (rPB) to control E1A and E1B gene expression Citation[52–54]. Two of these viruses, CV706 (originally named CN706) and CV787 have entered phase I clinical trials for prostate cancer. CV706 contains the E1A gene under control of the PSE sequence (the minimal PSA promoter together with the upstream PSA enhancer), and it is E3-deleted Citation[52]. Combination therapy using oncolytic CV706 and radiation therapy of prostate adenocarcinoma cells, LNCaP, resulted in synergistic cell killing both in vitro and in vivo Citation[55]. The combination therapy was not associated with increased toxicity and it was found that 50-fold less CV706 was needed to achieve complete tumor regression in the animal model when combined with radiation. The phase I clinical trial used intraprostatic injection of CV706 in patients relapsing after radiation therapy, as characterized by increased PSA. The therapy was demonstrated to be safe and serum PSA levels dropped ≥50% in the five patients receiving the highest dose of virus (1×1013 particles) Citation[56]. CV787 contains the E1A gene under control of the rPB promoter and the E1B gene under control of PSE, as well as the wild-type E3 region Citation[53]. Insertion of E3, which encodes proteins that assist virus release and play a role in evading or slowing host immune responses to the virus, enabled CV787 to eliminate distant LNCaP prostate cancer xenografts Citation[53]. The phase I clinical trial using intravenous delivery of CV787 has not yet been completed, although clinical safety of CV787 was reported at the 7th Annual American Society of Gene Therapy Meeting in June 2004. Final data from this trail are awaited.
In addition to prostate-specific promoters, the human osteocalcin promoter and the human telomerase reverse transcriptase (hTERT) promoter have been used to control E1A expression in CRAds with demonstrated effect in prostate cancer cells Citation[57], Citation[58]. Osteocalcin expression is restricted to bone-associated tissues, brain, and prostate cancer specimens and osteocalcin promoter-regulated CRAds have great potential for treatment of prostate cancer bone metastases. Telomerase is a unique DNA polymerase whose major function is to catalyze the addition of telomeric repeats to the ends of chromosomes to maintain chromosomal integrity. Most normal adult somatic cells are devoid of telomerase, which leads to a progressive shortening of the telomeres and ultimately results in senescence and cell death. Telomerase reactivation is a critical step for tumorigenesis, allowing cancer cells to proliferate indefinitely. Therefore, hTERT promoter-regulated CRAds have great potential for the treatment of a broad spectrum of tumors, including prostate cancer.
Therapeutic gene expression from tumor- and tissue-specific promoters
Promoters and enhancers
In addition to prostate CRAds, a number of replication-defective adenoviral vectors have been developed with therapeutic gene expression under the control of prostate-specific regulatory elements. Examples are adenoviral vectors with transgene expression based on promoter and enhancer elements from the genes encoding PSA Citation[59–64], kallikrein-2 Citation[65], prostate specific membrane antigen (PSMA) Citation[66], osteocalcin Citation[67], mouse prostate secretory protein of 94 amino acids (PSP94) Citation[68], and the composite rat probasin promoter (ARR2PB) Citation[69–74]. To increase the strength of prostate promoters while retaining satisfactory prostate specificity, two-step transcriptional activation systems have been developed. Zhang et al. Citation[75] developed a system in which a prostate-specific regulatory sequence, based on PSA promoter and enhancer elements, controls expression of a fusion protein comprising the DNA binding domain of the yeast transcription factor GAL4, and two transactivation domains of the Herpes simplex virus activator VP16. The produced fusion protein binds to a promoter, composed of GAL4 binding sites, which drives transgene expression. This system leads to higher transgene expression than a system where transgene expression is directly controlled by the prostate-specific regulatory sequence. Yoshimura et al. Citation[76] combined the PSA promoter and the bacteriophage P1 Cre-loxP system to generate a new prostate-specific transcription system. PSA-dependent expression of the Cre recombinase triggers the excision of a stop codon flanked by a pair of loxP sequences which releases transgene expression from the strong cytomegalovirus (CMV) promoter.
Since prostate cancer patients are often treated by androgen withdrawal, it may be beneficial to the patient to use a regulatory sequence that results in high therapeutic gene expression even in the absence of testosterone. Therefore, combining different prostate promoters and enhancers with different characteristics may be advantageous to treat both hormone-dependent and hormone-refractory prostate cancer. Lee et al. Citation[66] constructed an artificial prostate-specific enhancing sequence (PSES) comprising a minimal androgen-responsive PSA enhancer element and a minimal PSMA enhancer element (from the third intron) linked to a TATA-box promoter. When inserted into an adenoviral vector the PSES sequence showed prostate specificity and androgen independence both in vitro and in vivo. The activity of PSES in androgen-deprived cells comes from the PSMA enhancer that exerts increased activity in response to testosterone removal. Martiniello-Wilks et al. Citation[77] constructed an adenovirus with an artificial regulatory sequence comprising the rPB promoter and the simian virus 40 (SV-40) enhancer. The artificial regulatory sequence conferred prostate-specific transgene expression with activity even in the absence of testosterone. Furuhata et al. Citation[78] modified the androgen response elements (ARE) in the rPB promoter to construct a regulatory sequence that is induced by retinoids rather than by testosterone. Recently, we cloned the promoter for a prostate-specific transcript from the unrearranged T cell receptor γ-chain (TCRγ) locus, encoding the TCRg alternate reading frame protein (TARP) Citation[79]. An adenoviral vector with a recombinant regulatory sequence comprising the TARP promoter, the PSMA enhancer, and the PSA enhancer (named PPT) was constructed Citation[80]. In order to obtain absolute prostate specificity, a DNA insulator was introduced to shield the PPT sequence from adenoviral backbone sequences that otherwise interfere with transcriptional regulation. The insulator-shielded PPT adenovirus confers high prostate-specific gene expression in both the presence and absence of testosterone. Intravenous administration of the virus in nude mice with an orthotopic prostate tumor model revealed absolute tumor selectivity and a transcriptional activity fully comparable to the CMV promoter Citation[80]. The PPT sequence is currently being used to control E1A expression in an effort to produce highly active prostate-specific CRAds in our laboratory.
Therapeutic genes
The adenoviral E1A gene is a potent therapeutic gene in the sense that when controlled by a tumor- or tissue-specific promoter, it restricts viral replication and oncolysis of cells where the promoter is active. Besides E1A, a number of genes encoding various therapeutic molecules have been utilized for adenoviral-based prostate cancer gene therapy. Examples are adenoviral vectors with prostate-specific expression of prodrug-activating enzymes, also referred to as suicide genes, such as herpes simplex virus thymidine kinase (HSV-TK) Citation[60], Citation[78], Citation[81], Escherichia coli cytosine deaminase (CD) Citation[76], and Escherichia coli nitroreductase Citation[61]. HSV-TK facilitates the conversion of the prodrug ganciclovir (GCV) to the toxic GCV triphosphate, CD catalyzes the conversion of the nontoxic prodrug 5-fluorocytosine (5-FC) to the cytotoxic 5-fluorouracil (5-FU) and nitroreductase activates the prodrug CB1954 to a toxic metabolite. Other examples are adenoviral vectors with prostate-specific expression of apoptotic molecules such as Bax Citation[69], Citation[70], Citation[72], Citation[74], Bad Citation[72], caspase-8 Citation[82], and caspase-9 Citation[65]. Yet other examples include adenoviral vectors with prostate-specific expression of diphtheria toxin A Citation[64] and the sodium iodine symporter (NIS), which pumps iodine into cells and has utility in both imaging and therapy when combined with radioiodine Citation[73]. Other molecules of interest that could be inserted in adenoviral vectors under prostate-specific expressional control include TRAIL, Fas Ligand, Caspase 3, and pseudomonas exotoxin, some of which have been developed in non-viral systems.
In addition to adenoviral vectors with prostate-specific transgene expression, numerous adenoviral vectors have been developed with suicide genes, apoptotic genes, tumor suppressor genes, or immunostimulatory genes under control of strong ubiquitous promoters, such as the CMV promoter. Many such viral vectors have been used for experimental and clinical prostate cancer therapy Citation[83–85].
Restricting adenoviral replication to tumor cells using viral gene mutants
A number of genetically modified adenoviruses that preferentially replicate in tumor cells have been developed. The dl1520 strain (also called ONYX-015) contains alterations in the E1B-55kD gene that result in complete abrogation of E1B-55kD protein expression Citation[86]. In wild-type adenovirus infection the E1B-55kD protein binds to p53 and blocks its apoptotic activity, thereby allowing efficient virus replication. The theory was that dl1520 would not be able to inactivate p53 in normal cells and, as a result, the viral replication cycle would not be completed. In cancer cells lacking normal p53 function, however, replication was predicted to proceed. This means that ONYX-015 should selectively kill cancer cells with mutated p53. However, several reports have indicated that ONYX-015 replication in tumor cells can be independent of the p53 status Citation[87], Citation[88]. The reason for this may be that functional inactivation of p53, caused by alterations in MDM-2 or p14ARF protein expression, can make the tumor cell permissive to viral replication. Other examples of adenoviruses with genetic modification are the Δ24 strain Citation[89] and the dl922-947 strain Citation[90]. They both have small deletions in conserved region 2 of E1A that mediate interaction with the pRb family of proteins, which regulate the G1- to S-phase cell cycle checkpoint. These viruses confer selective replication in cells with a disrupted pRb pathway. Neither p53 nor pRb is mutated at a high frequency in early invasive prostate carcinoma Citation[91], which may somewhat limit their use in the treatment of localized prostate cancer.
To improve therapeutic efficacy, Freytag and colleagues have constructed a replication-competent dl1520 adenovirus strain (FRG, also called Ad5-CD/Tkrep) that, in addition to the lack of E1B-55kD protein expression, encodes a fusion protein comprising the CD and HSV-TK prodrug converting enzymes Citation[92]. They found that both the CD/5-FC and HSV-TK/GCV systems enhanced the cytopathic effect of the virus. The greatest therapeutic effect was seen in the virus plus double suicide gene therapy group compared with virus alone or with virus plus single suicide gene groups, suggesting synergistic interactions between the two suicide gene/prodrug systems Citation[92], Citation[93]. Moreover, a trimodal approach with Ad5-CD/TKrep, involving cell lysis by the replicating virus, double suicide gene therapy, and radiotherapy, led to significantly enhanced therapeutic efficacy compared with external beam radiotherapy alone in an orthotopic mouse prostate cancer model Citation[94]. Both suicide gene systems can be used to suppress viral replication, thereby providing means to control viral spread, but at the same time dampen the lytic effect of the virus in cancer cells. Therefore, it is important to examine other suicide gene systems in combination with oncolytic adenoviruses that possess primarily anti-tumor and not anti-viral action.
The Ad5-CD/TKrep replicating adenovirus has recently entered phase I clinical trials for localized prostate cancer. It is used in combination with double suicide gene therapy (5-FC and GCV) and conventional-dose three-dimensional conformal radiation therapy Citation[95], Citation[96]. The safety of the trimodal therapy is good with a toxicity profile that does not differ from radiation therapy alone Citation[96]. There are indications that the PSA level is declining much faster in the trimodal therapy group than in the group receiving radiotherapy alone. Furthermore, the number of negative biopsies after trimodal therapy is higher than expected for radiation therapy, suggesting possible interactions between the oncolytic adenovirus therapy, the double-suicide gene therapies and the radiation therapy, as reported at the 7th Annual American Society of Gene Therapy Meeting in June 2004.
Retargeted adenovirus infection
Three distinct sequential steps are required for adenoviral infection. These involve attachment of the adenovirus to a specific receptor on the surface of the target cell, internalization of the virus and transfer of the viral genome to the nucleus where it can be expressed. The primary virus to cell interaction of type 5 adenovirus occurs between the knob domain of the viral fiber and the cell surface Coxsackie/adenovirus receptor (CAR). CAR is strictly a docking site for the adenovirus, providing a high affinity virus to host association. The second interaction occurs between the RGD (Arg-Gly-Asp) amino acid motif of the viral penton base with the cellular integrins αvβ3 and αvβ5. This interaction triggers internalization of the virions through clathrin-mediated endocytosis and acidification of the endosomes, which allows the virions to escape and enter the cytosol. Following escape from the endosomes the partially dismantled virus is transported to the nuclear membrane and the virus genome is released into the nucleoplasm through nuclear pore complexes. Therefore, the capability of an adenovirus type 5 vector to infect a cell is based on CAR and integrin expression, and cells expressing CAR and integrins below a certain threshold level are refractory to adenovirus infection. It has been observed that many primary tumor cells and tumor cell lines are indeed refractory to adenovirus infection due to a deficiency of CAR expression. However, in the case of clinical prostate cancer, CAR expression is relatively well maintained Citation[97], Citation[98], which is advantageous for adenovirus-based prostate cancer gene therapy.
In order to retarget adenovirus infection to specific cells types, two general approaches have been utilized. One approach involves conjugate-based strategies, which require that the adenovirus is complexed with a targeting molecule that recognizes a specific cell surface receptor. A second approach is genetic retargeting, in which the virus fiber or capsid is genetically modified to recognize a specific cell surface receptor.
Conjugate-based adenovirus retargeting
Several strategies have been described to achieve functional linkage between the adenovirus fiber and a cell surface receptor. For example, a bi-specific antibody conjugate was used to target adenovirus to the epidermal growth factor (EGF) receptor Citation[99]. In an alternative approach, a single chain Fv antibody specific for the adenovirus fiber knob was genetically fused to EGF Citation[100]. The recombinant fusion protein was able to retarget adenovirus to the EGF receptor. Prostate cancer cells generally show abundant expression of EGF receptors Citation[101] and adenoviruses retargeted to EGF receptors may be utilized for enhanced adenovirus targeting to prostate cancer cells. So far, no paper has been published where an adenovirus was successfully retargeted to a prostate-specific cell surface molecule, although there are several known candidate molecules expressed on the surface of prostate cancer cells, including PSMA Citation[14], PSCA Citation[17], STEAP Citation[22], PSGR Citation[28], and NGEP Citation[40]. Cell surface molecules with potential use for prostate-specific retargeting of adenoviruses were recently reviewed by Maitland et al. Citation[102].
Genetic modification of the adenovirus fiber or capsid
The adenovirus fiber knob may be genetically reengineered to bind to a specific cell surface molecule. Wickham et al. Citation[103] constructed an adenovirus that contains a heparin-binding domain at the C-terminus of the fiber protein, which targeted the virus to broadly expressed heparan-containing cellular receptors. Dmitriev et al. Citation[104] reported the construction of modified adenoviral vectors containing the RGD (Arg-Gly-Asp)] peptide in the HI loop rather than at the C-terminus of the fiber domain. Attempts to retarget adenoviral vectors have also included genetic modifications of the penton base or hexon capsid proteins, although these proteins have not been exploited to the same extent as fiber-based targeting strategies. Hexon proteins are abundant on the adenovirus capsid and they contain hyper-variable regions that can be engineered to include targeting ligands. Vigne et al. Citation[105] modified the hyper-variable domain of the hexon proteins with a RGD peptide and found that the modified virus significantly increased the transduction of human vascular smooth muscle cells in vitro. Insertions of a targeting ligand into the knob domain have led to the expansion of viral tropism but not necessarily the ablation of CAR binding.
Fiber genes from other adenovirus serotypes can replace the native fiber gene in adenovirus type 2 and 5 vectors. This was first demonstrated in a publication by Gall et al. Citation[106] where removal of the adenovirus type 5 fiber and subsequent substitution with the adenovirus type 7 fiber resulted in altered viral tropism. It has been shown that substitution of the type 5 fiber with the type 35 fiber increases the adenovirus transduction efficiency of cells of hematopoietic origin, such as CD34+ stem cells and dendritic cells Citation[107], Citation[108]. Replacement of only the knob domain of the fiber can also alter viral tropism. This was first demonstrated by Krasnykh et al. Citation[109], who replaced the adenovirus type 5 knob with the type 3 knob.
Routes of adenovirus administration and immune responses
Initial clinical trials used intratumoral injections of replication-defective or CRAd adenoviruses in order to maximize safety. More recently systemic administration routes have been utilized, for example intravenous injection of ONYX-015 virus for the treatment of head and neck cancer Citation[110]. Clinical trials using systemic administration of adenovirus appear to be safe and the treatment well tolerated as long as lower doses are injected Citation[111]. However, systemic administration faces problems of systemic immune responses in the form of innate immunity and neutralizing antibodies. The innate response, which occurs within 24 hours, is dose-dependent and independent of viral transcription. It is directed against the viral particle and results in inflammation of transduced tissues. Ongoing clinical trials with single or repeated systemic administration of adenovirus will hopefully enhance our understanding of adenoviral pharmacokinetics and anti-adenoviral immune responses.
Adenoviruses also induce adaptive antiviral immunity, which can either inhibit or enhance the intended therapeutic effect. In the case of CRAd viruses, it is desirable to obtain maximal virus spread within the tumor before the virus is cleared by the host immune system. Therefore, insertion of the adenovirus E3 region into the virus vector, in full or in part, may help to slow down immune responses to the virus. The E3 gp19K protein binds to MHC class I molecules and prevents the export of MHC class I/antigenic peptide complexes to the cell surface, thereby blocking the killing of infected cells by cytolytic T lymphocytes. The E3 RID and E3 14.7K proteins also help to blunt the immune response by inhibiting tumor necrosis factor (TNF)-mediated apoptosis. The E3 11.6K protein named adenovirus death protein ensures efficient release of progeny virus from infected cells. CRAd viruses, which overexpress the adenovirus death protein, have increased cell lysis capacity and this leads to improved adenoviral spread Citation[112]. The intentional tuning of the antiviral immune response in adenoviral-based therapies is delicate. A delayed immune response ensures efficient spreading of CRAd viruses. On the other hand, a strong antiviral immune response may activate cytolytic T lymphocytes against tumor-associated antigens, which may lead to a desirable immunologic bystander effect with enhanced destruction of tumor cells.
Concluding remarks and future directions
Extensive research focusing on adenoviral vectors has allowed for significant advances in the field of cancer gene therapy. The field is rapidly moving from replication-defective to replication-competent adenoviral vectors. CRAd viruses offer two appealing properties as agents for cancer therapy: they specifically kill targeted cells and their replication leads to amplification of the therapeutic effect. Intraprostatic and intravenous injections of oncolytic CRAds have shown good safety profiles and encouraging potential when combined with external radiation therapy. In addition, hundreds of patients have been injected with various oncolytic adenoviruses, Herpes simplex viruses, or Newcastle disease viruses in a large number of clinical trials and without severe toxicity or side effects. Therefore, the focus on CRAd development is moving from primary concerns about safety towards how to make these viruses more efficient by increasing virus replication and virus spread within the tumor. Preclinical data suggest that efficacy may be improved by arming CRAds with therapeutic genes. It may be equally important to include genes encoding molecules for immune system modulation into the CRAds. Genes encoding molecules that suppress the immune response against the adenovirus may increase virus spread and thereby enhance the oncolytic effects. However, the host immune system will eventually clear the CRAds viruses and when this happens it might be advantageous to have genes encoding molecules that direct the immune response to a T helper 1 [TH1] cell-type response, in order to activate cytolytic T lymphocytes against tumor-associated antigens and thereby obtain an immunologic bystander effect.
Immunotherapy
A central question in tumor immunology is whether recognition of tumor-associated antigens (TAAs) by the immune system leads to activation or tolerance. Evidence that T lymphocytes can help to control tumor growth is provided by the analysis of tumor prevalence in immunodeficient individuals. The incidence of tumors of viral origin is increased markedly in patients with drug-induced or HIV-related immunodeficiency Citation[113]. However, for human tumors of non-viral origin, the incidence is similar in immunodeficient and healthy individuals, suggesting that for most human tumors no significant pressure is exerted by naturally occurring tumor immunity. If a specific immune surveillance system operates at early stages of tumorigenesis, it has most likely been eluded by the tumor at the time of tissue invasion and metastasis. It is therefore plausible that most tumors induce tolerance at an early stage of development and that advanced tumors may be particularly resistant to immunotherapies, as tolerance may be firmly established.
However, a number of important discoveries over the past two decades have led to renewed enthusiasm for cancer immunotherapy. First, the identification of human TAAs that can be recognized by autologous T cells, a finding that was first reported by Boon and colleagues Citation[114], Citation[115]. Second, discoveries were made showing that professional antigen-presenting cells must be activated in order to induce effective T cell immunity and that pattern recognition receptors and danger signals link innate to adoptive immunity via dendritic cells [DCs] Citation[116], Citation[117]. Third, the discovery that regulatory T lymphocytes, which are important for maintenance of immunological self-tolerance, are also involved in the induction of peripheral tolerance to TAAs and that depletion of these regulatory T lymphocytes can abrogate immunological unresponsiveness to tumors in vivo Citation[118–121].
Cancer immunotherapy through T cell activation
Early attempts at cancer immunotherapy focused on interleukin (IL)-2 or other T cell-activating cytokines that were intended to expand the number and potency of T cells reactive with TAAs. Such activated T cells were expected to infiltrate the tumor and lead to the specific destruction of tumor cells. In addition, early attempts were carried out with tumor cell lysate or irradiated tumor cells that were either mixed with adjuvants or genetically modified to increase immunogenicity. Although these attempts to boost anti-tumor immunity have yielded some limited examples of tumor regression, they have failed so far to control advanced-stage disease to a significant extent Citation[122]. As we learned more about antigen presentation, vaccination strategies were developed where peptide antigens or plasmids encoding TAAs, emulsified in immune adjuvants, were injected directly into patients, typically intramuscularly. Clinical trials using such vaccination strategies have only rarely and sporadically led to regressions of established tumors in patients with metastatic disease Citation[123].
The low clinical response rates may partly be due to the fact that the tumor environment lacks the danger signals that link innate to adoptive immunity, which is central for the immune system in eradicating foreign pathogens. The lack of danger may prevent the development of a general inflammatory reaction and may account for the failure of antigen-specific T cells to eliminate antigen-expressing tumor cells in vivo. Pattern recognition receptors such as the toll-like receptors (TLRs) link innate to adaptive immunity. Therefore, these receptors may be targeted to develop cancer immunotherapy strategies that mimic the efficient immune responses raised against foreign pathogens. For example, oligodeoxynucleotides (ODNs) with unmethylated CpG motifs, abundant in viral and prokaryotic genomes, can trigger an immunomodulatory cascade in humans which involves B cells, T cells, natural killer cells and DCs Citation[124]. The immune response triggered by CpG ODNs, via TLR-9 signaling, skews the immune system in favor of a TH1 cell-type response and pro-inflammatory cytokine production. The inclusion of CpG ODNs as vaccine adjuvants has shown promising results in some animal models Citation[125]. Double-stranded RNA (dsRNA), a major viral signature, activates DCs via TLR-3 or cytosolic dsRNA-binding enzyme protein kinase R. This activation leads to secretion of IL-12 and type-1 interferons which are important cytokines linking innate and adaptive immunity. An alphavirus replicon, which is a major source of dsRNA, or poly(I:C), a synthetic analog of dsRNA, may therefore be used to increase danger in the tumor environment. In addition to the lack of danger signaling, it will be important to overcome the escape mechanisms of tumors, including the loss of antigen expression and antigen presentation by tumor cells, the local presence of immunosuppressive factors or cells in the tumors, and the tumor-induced inability of lymphocytes to home to and infiltrate tumor tissues.
Dendritic cell vaccines
The ultimate effector cell that mediates the rejection of solid tumors in preclinical animal models is the cytolytic T lymphocyte (CTL). CTLs are derived from naïve or memory CD8+ T cells. Each CTL expresses a clonotypically unique T cell receptor (TCR) that confers specificity for a particular target antigen. The antigens recognized by TCRs on CTLs consist of peptide fragments bound within the major histocompatibility complex (MHC) class I molecules on the surface of the antigen-presenting cell. The peptides displayed by MHC class I molecules are generally from endogenous proteins expressed by the antigen presenting cell. DCs are specialized professional antigen presenting cells that are very efficient in presenting antigenic peptides to T cells along with co-stimulatory molecules needed for T cell activation. They exist as two distinct phenotypes, immature DCs specialized in antigen uptake and mature DCs specialized in antigen presentation. DCs have the unique capacity to stimulate naïve T cells and thereby initiate primary immune responses Citation[126]. Therefore, in order to initiate anti-tumor CTL responses, peptides derived from TAAs must be presented by MHC class I molecules on activated DCs.
Immature DCs can readily be generated from peripheral blood monocytes with IL-4 and granulocyte macrophage colony stimulating factor (GM-CSF). They may then be matured with tumor necrosis factor (TNF)-a or other maturation stimuli. Immature DCs can be loaded with the full-length TAA in the form of a recombinant protein, in vitro transcribed mRNA encoding the antigen, or a viral vector comprising the cDNA encoding the antigen. DCs are then matured for optimal processing and presentation of immunogenic peptide epitopes. Alternatively, fully mature DCs can be pulsed with synthetic MHC class I-restricted peptides for direct presentation of known immunogenic peptide epitopes. Autologous antigen-modified mature DCs can be infused as a vaccine into cancer patients Citation[127]. Clinical trials using DC vaccines against established tumors show that they are safe, with minimal side effects Citation[128]. The clinical results have been variable with notably long-lasting clinical responses in some studies Citation[128]. However, several of the initial vaccines used immature rather than mature DCs, which may have affected the immunological and clinical outcome. Only mature DCs have the ability to stimulate T cell responses and enhance homing to draining lymph nodes, the site where therapeutic T cell responses are initiated.
Adoptive transfer of tumor antigen-specific T lymphocytes
One interesting approach that may improve therapeutic outcome is to expand CTLs directed against TAAs ex vivo and adoptively transfer them back to the cancer patient. In such a therapeutic setting lymphocyte cultures are repeatedly stimulated with autologous DCs modified to present immunogenic TAA epitopes. Specific CTLs with high avidity for an antigenic epitope can then be identified by peptide/MHC tetramer or enzyme-linked immunosorbent assay (ELISA) technologies. T cell clones can be raised from a single T cell, or a specific T cell population can be isolated by fluorescent-activated cell sorting (FACS) using a peptide/MHC tetramer. Isolated lymphocyte cultures can be rapidly expanded ex vivo, to clinically relevant numbers for adoptive transfer back to the patient (see ). Alternatively, the molecular characterization of TAA epitopes and the TCRs that recognize them has opened up the possibility to genetically engineer autologous T cells ex vivo to express a full-length TCR or a chimeric single chain Fv receptor that is specific for a TAA epitope Citation[129].
Figure 3. Adoptive transfer of prostate antigen-specific T cells. Monocyte-derived dendritic cells (DCs) are genetically modified with a prostate antigen-encoding adenovirus vector. The modified DCs are mixed with autologous lymphocytes for stimulation of antigen-specific T cells ex vivo. Antigen-specific T cells are selected and rapidly expanded to large numbers for adoptive therapy. The patient receives systemic immunosuppressive chemotherapy prior to the adoptive transfer of T cells.
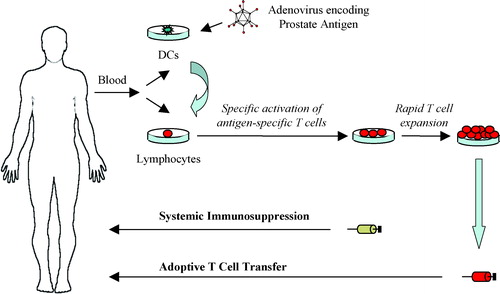
In contrast with early studies of adoptive therapy that used lymphokine-activated killer (LAK) cells or tumor infiltrating lymphocytes (TILs), the use of antigen-specific T cell clones for adoptive transfer provides effector cells of uniform specificity and phenotype that retain physiological responses to antigen stimulation and low-dose IL-2 Citation[130]. Very large numbers of highly selected cells can be adoptively transferred. One important aspect of adoptive T cell transfer is that the T cells are expanded ex vivo, thereby circumventing the host immune regulatory mechanisms and the potentially immunosuppressive tumor environment. Another important aspect is that the patient can receive systemic immunosuppressive chemotherapy before the T cell transfer. This lymphodepletion creates space and provides homeostatic lymphocyte survival and proliferative signals for the adoptively transferred T cells Citation[131]. Moreover, it has been suggested that the success of adoptive T cell transfer for the treatment of Epstein–Barr virus (EBV)-related lymphoma and for prophylaxis of CMV infection is due, in part, to the immunosuppressed status of the treated patients. Therefore, it may well be that lymphodepletion prior to adoptive transfer of TAA-specific CTLs can help to induce the inflammatory response that is needed for the CTLs to exert their cytolytic action.
It is well established that CD25+ CD4+ regulatory T cells (Treg) are important for the maintenance of immunological tolerance. They have also been found to have a profound effect on the inhibition of T cell responses against cancer Citation[118]. Systemic depletion of Treg cells with high dose anti-CD25 antibody or immunotoxin prior to adoptive transfer of antigen-specific T cells may have implications for cancer immunotherapy. However, activated effector CD8+ and CD4+ cells also express CD25, and depletion of these cells during the acute phase of antitumor T cell response may limit the use of this approach. Moreover, it creates a greater risk for autoimmunity than a general lymphodepletion, since the potentially autoreactive T cells would not be depleted along with the Treg cells. It has recently been shown that TAA-specific CD25+ CD4+ Treg cells are present at tumor sites Citation[132]. General application of Treg cell depletion in the clinics will probably await methods to preferentially deplete Treg cell subsets that regulate antitumor responses.
Prostate cancer immunotherapy
There are two main reasons why prostate cancer is particularly well suited to immunotherapy and adoptive T cell therapy. First, prostate tissues express many unique antigens, which may be exploited for the induction of anti-tumor CTL responses. Prostate cancer cells also express many epithelial cancer-related antigens that may be exploited as well. Moreover, immune responses to prostate-related antigens can be considered tumor-specific in patients with recurrent metastases after radical prostatectomy. Second, detection of serum PSA is available as an evaluation marker of clinical responses.
A fair number of immunogenic peptides derived from the prostate tissue antigens PSA Citation[133–137], PSMA Citation[138–142], PAP Citation[143], Citation[144], PSCA Citation[145–147], kallikrein 4 [hK-4], prostein Citation[148], Citation[149], trp-p8 Citation[150], and TARP Citation[151], Citation[152] have been identified in vitro. They are restricted by HLA-A2, HLA-A3, or HLA-A24, which are all relatively common MHC class I molecules in the general population. MHC class II-binding peptides that can activate CD4+ T helper cells have so far been identified from PSA Citation[153], PSMA Citation[153–155], PAP Citation[156], and hK4 Citation[157]. T helper cell activation is essential for a sustained immunological response against tumor cells. In addition, our laboratory has recently shown that potent CTLs directed against the prostate tissue antigen TARP can be generated ex vivo by repeated stimulation of lymphocyte cultures with autologous peptide-pulsed DCs followed by peptide/MHC tetramer-guided sorting of the stimulated T cells and 1000-fold rapid expansion of the sorted T cells Citation[152]. The sorted and expanded T cells retained both specificity and activity, demonstrating that large numbers of prostate tissue antigen-specific CTLs can be generated ex vivo for adoptive transfer to prostate cancer patients Citation[152].
Several phase I and phase II clinical trials have been conducted with autologous DCs preloaded with prostate tissue antigens. Murphy and colleagues were first, using DCs pulsed with HLA-A2-restricted PSMA peptides Citation[141], Citation[158], Citation[159]. Burch et al. Citation[160] and Small et al. Citation[161] used DCs pulsed with a recombinant fusion protein composed of PAP linked to GM-CSF, while Fong et al. Citation[162] used DCs pulsed with murine PAP as a xenoantigen. Heiser et al. Citation[163] used DCs transfected with mRNA encoding PSA while Barrou et al. Citation[164] used DCs pulsed with recombinant PSA. Immunotherapy using antigen-presenting cells (APC8015, Provenge) pulsed with the recombinant PAP/GM-CSF fusion protein has been conducted as phase II Citation[165] and phase III (not yet published) clinical trials for metastatic androgen-independent prostate cancer with sporadic although encouraging results. Generally speaking, the DC vaccine trials for prostate cancer have been safe and well tolerated by the patients. Immunologic responses have been limited but specific T cell responses were observed in certain patients, demonstrating that it is possible to activate CTLs against self-antigens such as PSA, PSMA, and PAP in vivo in a clinical setting.
As mentioned above, telomerase reactivation is a critical step for tumorigenesis. Therefore, peptides derived from hTERT may be used to expand T cells with activity against a broad spectrum of tumors, including prostate cancer. hTERT peptides restricted by HLA-A2 Citation[166], Citation[167], HLA-A3 Citation[168], and HLA-A24 Citation[169] have been identified, as well as various MHC class II-restricted hTERT peptides Citation[170], Citation[171]. Vonderheide et al. Citation[172] recently performed a phase I clinical vaccination trial with autologous dendritic cells pulsed with an HLA-A2-restricted hTERT peptide. hTERT-specific T lymphocytes were induced in four out of seven patients with advanced breast or prostate carcinoma, as assessed by peptide/MHC tetramer, enzyme-linked immunospot, and cytotoxicity assays.
So far, no clinical trial has been conducted with adoptive transfer of prostate antigen-specific T cells to prostate cancer patients.
Concluding remarks and future directions
Immunotherapy of cancer is a field of extensive research activity and recent discoveries in tumor immunology have renewed hopes that immunotherapy may play a role in the treatment of clinical cancers. So far, the clinical results have not fulfilled expectations and only sporadic tumor regressions have been documented. The main problem that remains to be solved is how to overcome the immunological tolerance that appears to be firmly established in most advanced tumors. One interesting approach that is being explored is ex vivo expansion and adoptive transfer of autologous TAA-specific CTLs to cancer patients who have received systemic immunosuppression prior to the adoptive T cell therapy. Growing knowledge about lymphocyte characteristics, other than antigen specificity, that can impact lymphocyte behavior in vivo may be utilized for the design of improved adoptive therapy protocols. For example, the discovery that the maturation state of CD8+ T cells determines their in vivo transport and persistence during an immune response Citation[173] opens up new possibilities to improve the behavior of T cells. However, adoptive T cell transfer is a treatment that requires advanced logistics. Therefore, vaccination strategies based on dendritic cells, tumor antigens, cytokines, and novel immune adjuvants such as CpG ODNs will be continuously explored. Considering the fact that a large proportion of prostate cancer is hereditary and that many prostate tissue antigens have been characterized, prophylactic prostate cancer vaccines may be explored as well.
General conclusions
Prostate cancer remains a leading cause of illness and death among men in Western countries. Early detection and aggressive treatment are essential parameters to improve the overall survival of patients, although this will lead to over-treatment for many individuals. In the case of localized prostate cancer, intraprostatic injection of oncolytic prostate CRAds has shown great potential when combined with radiation therapy. Furthermore, it is plausible to administer CRAds concomitant with radical prostatectomy in order to trace tumor cells that may have disseminated from the prostate capsule. In the case of metastatic prostate cancer the situation is far more complicated and no curative treatment exists today. Several studies have demonstrated that TAA-specific CTLs can be activated both in vitro and in vivo, clearly showing that the host immune system can be activated against tumors. As DC-based vaccination and adoptive T cell transfer strategies become more and more sophisticated, it is essential to include them early on in the course of recurrent prostate cancer, preferably along with the standard hormonal treatments. It is hoped thay this will lead to a role for immunotherapy in controlling prostate cancer by extending survival and improving the quality of life.
The author would like to thank Christina Ninalga, Helena Dzojic, Wing-Shing Cheng, Björn Carlsson, and Dr Valeria Giandomenico for critical reading of the paper.
References
- Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin 2003; 53: 5–26
- Jani AB, Hellman S. Early prostate cancer: clinical decision-making. Lancet 2003; 361: 1045–53
- Nilsson S, Norlen BJ, Widmark A. A systematic overview of radiation therapy effects in prostate cancer. Acta Oncol 2004; 43: 316–81
- Scattoni V, Montorsi F, Picchio M, Roscigno M, Salonia A, Rigatti P, Fazio F. Diagnosis of local recurrence after radical prostatectomy. BJU Int 2004; 93: 680–8
- Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351: 1513–20
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351: 1502–12
- Ryan CJ, Small EJ. Advances in prostate cancer. Curr Opin Oncol 2004; 16: 242–6
- Moul JW, Wu H, Sun L, McLeod DG, Amling C, Lance R, et al. Epidemiology of radical prostatectomy for localized prostate cancer in the era of prostate-specific antigen:an overview of the Department of Defense Center for Prostate Disease Research national database. Surgery 2002; 132: 213–19
- Stattin P, Johansson R, Damber JE, Hellstrom M, Hugosson J, Lundgren R, et al. Non-systematic screening for prostate cancer in Sweden: Survey from the National Prostate Cancer Registry. Scand J Urol Nephrol 2003; 37: 461–5
- Johansson JE, Andren O, Andersson SO, Dickman PW, Holmberg L, Magnuson A, Adami HO. Natural history of early, localized prostate cancer. JAMA 2004; 291: 2713–9
- Lundwall A, Lilja H. Molecular cloning of human prostate specific antigen cDNA. FEBS Lett 1987; 214: 317–22
- Schedlich LJ, Bennetts BH, Morris BJ. Primary structure of a human glandular kallikrein gene. DNA 1987; 6: 429–37
- Yeh LC, Lee AJ, Lee NE, Lam KW, Lee JC. Molecular cloning of cDNA for human prostatic acid phosphatase. Gene 1987; 60: 191–6
- Israeli RS, Powell CT, Corr JG, Fair WR, Heston WD. Expression of the prostate-specific membrane antigen. Cancer Res 1994; 54: 1807–11
- Grant FJ, Taylor DA, Sheppard PO, Mathewes SL, Lint W, Vanaja E, et al. Molecular cloning and characterization of a novel transglutaminase cDNA from a human prostate cDNA library. Biochem Biophys Res Commun 1994; 203: 1117–23
- He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics 1997; 43: 69–77
- Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen:a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA 1998; 95: 1735–40
- Brinkmann U, Vasmatzis G, Lee B, Yerushalmi N, Essand M, Pastan I. PAGE-1, an X chromosome-linked GAGE-like gene that is expressed in normal and neoplastic prostate, testis, and uterus. Proc Natl Acad Sci USA 1998; 95: 10757–62
- Essand M, Vasmatzis G, Brinkmann U, Duray P, Lee B, Pastan I. High expression of a specific T-cell receptor gamma transcript in epithelial cells of the prostate. Proc Natl Acad Sci USA 1999; 96: 9287–92
- Wolfgang CD, Essand M, Lee B, Pastan I. T-cell receptor gamma chain alternate reading frame protein (TARP) expression in prostate cancer cells leads to an increased growth rate and induction of caveolins and amphiregulin. Cancer Res 2001; 61: 8122–6
- Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, et al. DD3:a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res 1999; 59: 5975–9
- Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci USA 1999; 96: 14523–8
- Nelson PS, Gan L, Ferguson C, Moss P, Gelinas R, Hood L, Wang K. Molecular cloning and characterization of prostase, an androgen-regulated serine protease with prostate-restricted expression. Proc Natl Acad Sci USA 1999; 96: 3114–9
- Oettgen P, Finger E, Sun Z, Akbarali Y, Thamrongsak U, Boltax J, et al. PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem 2000; 275: 1216–25
- Nozawa M, Yomogida K, Kanno N, Nonomura N, Miki T, Okuyama A, et al. Prostate-specific transcription factor hPSE is translated only in normal prostate epithelial cells. Cancer Res 2000; 60: 1348–52
- Srikantan V, Zou Z, Petrovics G, Xu L, Augustus M, Davis L, et al. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc Natl Acad Sci USA 2000; 97: 12216–21
- Lin B, White JT, Ferguson C, Bumgarner R, Friedman C, Trask B, et al. PART-1:a novel human prostate-specific, androgen-regulated gene that maps to chromosome 5q12. Cancer Res 2000; 60: 858–63
- Xu LL, Stackhouse BG, Florence K, Zhang W, Shanmugam N, Sesterhenn IA, et al. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res 2000; 60: 6568–72
- Xu LL, Shanmugam N, Segawa T, Sesterhenn IA, McLeod DG, Moul JW, Srivastava S. A novel androgen-regulated gene, PMEPA1, located on chromosome 20q13 exhibits high level expression in prostate. Genomics 2000; 66: 257–63
- Xu J, Kalos M, Stolk JA, Zasloff EJ, Zhang X, Houghton RL, et al. Identification and characterization of prostein, a novel prostate-specific protein. Cancer Res 2001; 61: 1563–8
- Olsson P, Bera TK, Essand M, Kumar V, Duray P, Vincent J, Lee B, Pastan I. GDEP, a new gene differentially expressed in normal prostate and prostate cancer. Prostate 2001; 48: 231–41
- Tsavaler L, Shapero MH, Morkowski S, Laus R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res 2001; 61: 3760–9
- Lin B, White JT, Ferguson C, Wang S, Vessella R, Bumgarner R, et al. Prostate short-chain dehydrogenase reductase 1 (PSDR1): A new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res 2001; 61: 1611–18
- Lo SH, Lo TB. Cten, a COOH-terminal tensin-like protein with prostate restricted expression, is down-regulated in prostate cancer. Cancer Res 2002; 62: 4217–21
- Bera TK, Maitra R, Iavarone C, Salvatore G, Kumar V, Vincent JJ, et al. PATE, a gene expressed in prostate cancer, normal prostate, and testis, identified by a functional genomic approach. Proc Natl Acad Sci USA 2002; 99: 3058–63
- Bera TK, Zimonjic DB, Popescu NC, Sathyanarayana BK, Kumar V, Lee B, Pastan I. POTE, a highly homologous gene family located on numerous chromosomes and expressed in prostate, ovary, testis, placenta, and prostate cancer. Proc Natl Acad Sci USA 2002; 99: 16975–80
- Korkmaz KS, Elbi C, Korkmaz CG, Loda M, Hager GL, Saatcioglu F. Molecular cloning and characterization of STAMP1, a highly prostate-specific six transmembrane protein that is overexpressed in prostate cancer. J Biol Chem 2002; 277: 36689–96
- Qi H, Fillion C, Labrie Y, Grenier J, Fournier A, Berger L, et al. AIbZIP, a novel bZIP gene located on chromosome 1q21.3 that is highly expressed in prostate tumors and of which the expression is up-regulated by androgens in LNCaP human prostate cancer cells. Cancer Res 2002; 62: 721–33
- Herness EA, Naz RK. A novel human prostate-specific gene-1 (HPG-1): Molecular cloning, sequencing, and its potential involvement in prostate carcinogenesis. Cancer Res 2003; 63: 329–36
- Bera TK, Das S, Maeda H, Beers R, Wolfgang CD, Kumar V, et al. NGEP, a gene encoding a membrane protein detected only in prostate cancer and normal prostate. Proc Natl Acad Sci USA 2004; 101: 3059–64
- Wang R, Xu J, Saramaki O, Visakorpi T, Sutherland WM, Zhou J, et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Res 2004; 64: 1589–94
- Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy:the art of turning infectious agents into vehicles of therapeutics. Nat Med 2001; 7: 33–40
- Galanis E, Vile R, Russell SJ. Delivery systems intended for in vivo gene therapy of cancer:targeting and replication competent viral vectors. Crit Rev Oncol Hematol 2001; 38: 177–92
- Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat Biotechnol 2000; 18: 33–7
- Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene therapy clinical trials worldwide 1989–2004: An overview. J Gene Med 2004; 6: 597–602
- Mabjeesh NJ, Zhong H, Simons JW. Gene therapy of prostate cancer: Current and future directions. Endocr Relat Cancer 2002; 9: 115–39
- Palmer DH, Mautner V, Kerr DJ. Clinical experience with adenovirus in cancer therapy. Curr Opin Mol Ther 2002; 4: 423–34
- Chiocca EA, Aghi M, Fulci G. Viral therapy for glioblastoma. Cancer J 2003; 9: 167–79
- Edelman J, Nemunaitis J. Adenoviral p53 gene therapy in squamous cell cancer of the head and neck region. Curr Opin Mol Ther 2003; 5: 611–17
- Russell WC. Update on adenovirus and its vectors. J Gen Virol 2000; 81: 2573–604
- Morsy MA, Caskey CT. Expanded-capacity adenoviral vectors: The helper-dependent vectors. Mol Med Today 1999; 5: 18–24
- Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706:a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res 1997; 57: 2559–63
- Yu DC, Chen Y, Seng M, Dilley J, Henderson DR. The addition of adenovirus type 5 region E3 enables calydon virus 787 to eliminate distant prostate tumor xenografts. Cancer Res 1999; 59: 4200–3
- Yu DC, Sakamoto GT, Henderson DR. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer Res 1999; 59: 1498–504
- Chen Y, DeWeese T, Dilley J, Zhang Y, Li Y, Ramesh N, et al. CV706, a prostate cancer-specific adenovirus variant, in combination with radiotherapy produces synergistic antitumor efficacy without increasing toxicity. Cancer Res 2001; 61: 5453–60
- DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res 2001; 61: 7464–72
- Matsubara S, Wada Y, Gardner TA, Egawa M, Park MS, Hsieh CL, et al. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res 2001; 61: 6012–9
- Lanson NA, Jr, Friedlander PL, Schwarzenberger P, Kolls JK, Wang G. Replication of an adenoviral vector controlled by the human telomerase reverse transcriptase promoter causes tumor-selective tumor lysis. Cancer Res 2003; 63: 7936–41
- Gotoh A, Ko SC, Shirakawa T, Cheon J, Kao C, Miyamoto T, et al. Development of prostate-specific antigen promoter-based gene therapy for androgen-independent human prostate cancer. J Urol 1998; 160: 220–9
- Martiniello-Wilks R, Garcia-Aragon J, Daja MM, Russell P, Both GW, Molloy PL, et al. In vivo gene therapy for prostate cancer: Preclinical evaluation of two different enzyme-directed prodrug therapy systems delivered by identical adenovirus vectors. Hum Gene Ther 1998; 9: 1617–26
- Latham JP, Searle PF, Mautner V, James ND. Prostate-specific antigen promoter/enhancer driven gene therapy for prostate cancer: Construction and testing of a tissue-specific adenovirus vector. Cancer Res 2000; 60: 334–41
- Wu L, Matherly J, Smallwood A, Adams JY, Billick E, Belldegrun A, Carey M. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Ther 2001; 8: 1416–26
- Shi CX, Hitt M, Ng P, Graham FL. Superior tissue-specific expression from tyrosinase and prostate-specific antigen promoters/enhancers in helper-dependent compared with first-generation adenoviral vectors. Hum Gene Ther 2002; 13: 211–24
- Li Y, McCadden J, Ferrer F, Kruszewski M, Carducci M, Simons J, Rodriguez R. Prostatespecific expression of the diphtheria toxin A chain (DTA): Studies of inducibility and specificity of expression of prostate-specific antigen promoter-driven DT-A adenoviral-mediated gene transfer. Cancer Res 2002; 62: 2576–82
- Xie X, Zhao X, Liu Y, Young CY, Tindall DJ, Slawin KM, Spencer DM. Robust prostate-specific expression for targeted gene therapy based on the human kallikrein 2 promoter. Hum Gene Ther 2001; 12: 549–61
- Lee SJ, Kim HS, Yu R, Lee K, Gardner TA, Jung C, et al. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol Ther 2002; 6: 415–21
- Hsieh CL, Yang L, Miao L, Yeung F, Kao C, Yang H, et al. A novel targeting modality to enhance adenoviral replication by vitamin D(3) in androgen-independent human prostate cancer cells and tumors. Cancer Res 2002; 62: 3084–92
- Gabril MY, Onita T, Ji PG, Sakai H, Chan FL, Koropatnick J, et al. Prostate targeting: PSP94 gene promoter/enhancer region directed prostate tissue-specific expression in a transgenic mouse prostate cancer model. Gene Ther 2002; 9: 1589–99
- Andriani F, Nan B, Yu J, Li X, Weigel NL, McPhaul MJ, et al. Use of the probasin promoter ARR2PB to express Bax in androgen receptor-positive prostate cancer cells. J Natl Cancer Inst 2001; 93: 1314–24
- Lowe SL, Rubinchik S, Honda T, McDonnell TJ, Dong JY, Norris JS. Prostate-specific expression of Bax delivered by an adenoviral vector induces apoptosis in LNCaP prostate cancer cells. Gene Ther 2001; 8: 1363–71
- Rubinchik S, Wang D, Yu H, Fan F, Luo M, Norris JS, Dong JY. A complex adenovirus vector that delivers FASL-GFP with combined prostate-specific and tetracycline-regulated expression. Mol Ther 2001; 4: 416–26
- Zhang Y, Yu J, Unni E, Shao TC, Nan B, Snabboon T, et al. Monogene and polygene therapy for the treatment of experimental prostate cancers by use of apoptotic genes bax and bad driven by the prostate-specific promoter ARR(2)PB. Hum Gene Ther 2002; 13: 2051–64
- Kakinuma H, Bergert ER, Spitzweg C, Cheville JC, Lieber MM, Morris JC. Probasin promoter (ARR[2]PB)-driven, prostate-specific expression of the human sodium iodide symporter (h-NIS) for targeted radioiodine therapy of prostate cancer. Cancer Res 2003; 63: 7840–4
- Nan B, Snabboon T, Unni E, Yuan XJ, Whang YE, Marcelli M. The PTEN tumor suppressor is a negative modulator of androgen receptor transcriptional activity. J Mol Endocrinol 2003; 31: 169–83
- Zhang L, Adams JY, Billick E, Ilagan R, Iyer M, Le K, et al. Molecular engineering of a two-step transcription amplification (TSTA) system for transgene delivery in prostate cancer. Mol Ther 2002; 5: 223–32
- Yoshimura I, Ikegami S, Suzuki S, Tadakuma T, Hayakawa M. Adenovirus mediated prostate specific enzyme prodrug gene therapy using prostate specific antigen promoter enhanced by the Cre-loxP system. J Urol 2002; 168: 2659–64
- Martiniello-Wilks R, Tsatralis T, Russell P, Brookes DE, Zandvliet D, Lockett LJ, et al. Transcription-targeted gene therapy for androgen-independent prostate cancer. Cancer Gene Ther 2002; 9: 443–52
- Furuhata S, Ide H, Miura Y, Yoshida T, Aoki K. Development of a prostate-specific promoter for gene therapy against androgen-independent prostate cancer. Mol Ther 2003; 7: 366–74
- Cheng WS, Giandomenico V, Pastan I, Essand M. Characterization of the androgen-regulated prostate-specific T cell receptor gamma-chain alternate reading frame protein (TARP) promoter. Endocrinology 2003; 144: 3433–40
- Cheng WS, Kraaij R, Nilsson B, Van Der Weel L, De Ridder CM, Totterman TH, Essand M. A novel TARP-promoter-based adenovirus against hormone-dependent and hormone-refractory prostate cancer. Mol Ther 2004; 10: 355–64
- Kubo H, Gardner TA, Wada Y, Koeneman KS, Gotoh A, Yang L, et al. Phase I dose escalation clinical trial of adenovirus vector carrying osteocalcin promoter-driven herpes simplex virus thymidine kinase in localized and metastatic hormone-refractory prostate cancer. Hum Gene Ther 2003; 14: 227–41
- Komata T, Kondo Y, Kanzawa T, Ito H, Hirohata S, Koga S, et al. Caspase-8 gene therapy using the human telomerase reverse transcriptase promoter for malignant glioma cells. Hum Gene Ther 2002; 13: 1015–25
- Shalev M, Miles BJ, Thompson TC, Ayala G, Butler EB, Aguilar-Cordova E, Kadmon D. Suicide gene therapy for prostate cancer using a replication-deficient adenovirus containing the herpesvirus thymidine kinase gene. World J Urol 2000; 18: 125–9
- Norris JS, Hyer ML, Voelkel-Johnson C, Lowe SL, Rubinchik S, Dong JY. The use of Fas Ligand, TRAIL and Bax in gene therapy of prostate cancer. Curr Gene Ther 2001; 1: 123–36
- McNeish IA, Bell SJ, Lemoine NR. Gene therapy progress and prospects: Cancer gene therapy using tumour suppressor genes. Gene Ther 2004; 11: 497–503
- Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996; 274: 373–6
- Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol 1998; 72: 9470–8
- Goodrum FD, Ornelles DA. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol 1998; 72: 9479–90
- Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000; 19: 2–12
- Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med 2000; 6: 1134–9
- Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev 2000; 14: 2410–34
- Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: Concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther 1998; 9: 1323–33
- Rogulski KR, Wing MS, Paielli DL, Gilbert JD, Kim JH, Freytag SO. Double suicide gene therapy augments the antitumor activity of a replication-competent lytic adenovirus through enhanced cytotoxicity and radiosensitization. Hum Gene Ther 2000; 11: 67–76
- Freytag SO, Paielli D, Wing M, Rogulski K, Brown S, Kolozsvary A, et al. Efficacy and toxicity of replication-competent adenovirus-mediated double suicide gene therapy in combination with radiation therapy in an orthotopic mouse prostate cancer model. Int J Radiat Oncol Biol Phys 2002; 54: 873–85
- Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M, et al. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res 2002; 62: 4968–76
- Freytag SO, Stricker H, Pegg J, Paielli D, Pradhan DG, Peabody J, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res 2003; 63: 7497–506
- Pandha HS, Stockwin LH, Eaton J, Clarke IA, Dalgleish AG, Todryk SM, Blair GE. Coxsackie B and adenovirus receptor, integrin and major histocompatibility complex class I expression in human prostate cancer cell lines:implications for gene therapy strategies. Prostate Cancer Prostatic Dis 2003; 6: 6–11
- Rauen KA, Sudilovsky D, Le JL, Chew KL, Hann B, Weinberg V, et al. Expression of the coxsackie adenovirus receptor in normal prostate and in primary and metastatic prostate carcinoma: Potential relevance to gene therapy. Cancer Res 2002; 62: 3812–8
- Miller CR, Buchsbaum DJ, Reynolds PN, Douglas JT, Gillespie GY, Mayo MS, et al. Differential susceptibility of primary and established human glioma cells to adenovirus infection: Targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Res 1998; 58: 5738–48
- Watkins SJ, Mesyanzhinov VV, Kurochkina LP, Hawkins RE. The ‘adenobody’ approach to viral targeting: Specific and enhanced adenoviral gene delivery. Gene Ther 1997; 4: 1004–12
- Lorenzo GD, Bianco R, Tortora G, Ciardiello F. Involvement of growth factor receptors of the epidermal growth factor receptor family in prostate cancer development and progression to androgen independence. Clin Prostate Cancer 2003; 2: 50–7
- Maitland NJ, Stanbridge LJ, Dussupt V. Targeting gene therapy for prostate cancer. Curr Pharm Des 2004; 10: 531–55
- Wickham TJ, Roelvink PW, Brough DE, Kovesdi I. Adenovirus targeted to heparan-containing receptors increases its gene delivery efficiency to multiple cell types. Nat Biotechnol 1996; 14: 1570–3
- Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol 1998; 72: 9706–13
- Vigne E, Mahfouz I, Dedieu JF, Brie A, Perricaudet M, Yeh P. RGD inclusion in the hexon monomer provides adenovirus type 5-based vectors with a fiber knob-independent pathway for infection. J Virol 1999; 73: 5156–61
- Gall J, Kass-Eisler A, Leinwand L, Falck-Pedersen E. Adenovirus type 5 and 7 capsid chimera: Fiber replacement alters receptor tropism without affecting primary immune neutralization epitopes. J Virol 1996; 70: 2116–23
- Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A. Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J Virol 2000; 74: 2567–83
- Nilsson M, Karlsson S, Fan X. Functionally distinct subpopulations of cord blood CD34+ cells are transduced by adenoviral vectors with serotype 5 or 35 tropism. Mol Ther 2004; 9: 377–88
- Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J Virol 1996; 70: 6839–46
- Nemunaitis J, Cunningham C, Buchanan A, Blackburn A, Edelman G, Maples P, et al. Intravenous infusion of a replication-selective adenovirus (ONYX–015) in cancer patients: Safety, feasibility and biological activity. Gene Ther 2001; 8: 746–59
- Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: Lessons from clinical trials. Cancer Gene Ther 2002; 9: 979–86
- Doronin K, Toth K, Kuppuswamy M, Krajcsi P, Tollefson AE, Wold WS. Overexpression of the ADP (E3–11.6K) protein increases cell lysis and spread of adenovirus. Virology 2003; 305: 378–87
- Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Semin Oncol 2000; 27: 390–401
- Van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254: 1643–7
- Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev 2002; 188: 51–64
- Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 2002; 20: 621–67
- Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol 2001; 13: 114–19
- Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22: 531–62
- Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25 + CD4+ T cells: A common basis between tumor immunity and autoimmunity. J Immunol 1999; 163: 5211–8
- Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med 2001; 194: 823–32
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850–4
- Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature 2001; 411: 380–4
- Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998; 4: 321–7
- Klinman DM. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol 2004; 4: 249–58
- Davila E, Kennedy R, Celis E. Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG-oligodeoxynucleotide adjuvant, and CTLA-4 blockade. Cancer Res 2003; 63: 3281–8
- Mellman I, Steinman RM. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001; 106: 255–8
- Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: Mapping the way. Nat Med 2004; 10: 475–80
- Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest 2003; 21: 873–86
- Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol 2002; 2: 512–19
- Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma: Direct evidence of t cell-mediated vitiligo. J Exp Med 2000; 192: 1637–44
- Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer 2003; 3: 666–75
- Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, et al. Tumor–specific human CD4+ regulatory T cells and their ligands: Implications for immunotherapy. Immunity 2004; 20: 107–18
- Xue BH, Zhang Y, Sosman JA, Peace DJ. Induction of human cytotoxic T lymphocytes specific for prostate-specific antigen. Prostate 1997; 30: 73–8
- Correale P, Walmsley K, Nieroda C, Zaremba S, Zhu M, Schlom J, Tsang KY. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J Natl Cancer Inst 1997; 89: 293–300
- Correale P, Walmsley K, Zaremba S, Zhu M, Schlom J, Tsang KY. Generation of human cytolytic T lymphocyte lines directed against prostate-specific antigen (PSA) employing a PSA oligoepitope peptide. J Immunol 1998; 161: 3186–94
- Harada M, Kobayashi K, Matsueda S, Nakagawa M, Noguchi M, Itoh K. Prostate-specific antigen-derived epitopes capable of inducing cellular and humoral responses in HLA-A24+ prostate cancer patients. Prostate 2003; 57: 152–9
- Terasawa H, Tsang KY, Gulley J, Arlen P, Schlom J. Identification and characterization of a human agonist cytotoxic T-lymphocyte epitope of human prostate-specific antigen. Clin Cancer Res 2002; 8: 41–53
- Kobayashi K, Noguchi M, Itoh K, Harada M. Identification of a prostate-specific membrane antigen-derived peptide capable of eliciting both cellular and humoral immune responses in HLA-A24+ prostate cancer patients. Cancer Sci 2003; 94: 622–7
- Horiguchi Y, Nukaya I, Okazawa K, Kawashima I, Fikes J, Sette A, et al. Screening of HLA-A24-restricted epitope peptides from prostate-specific membrane antigen that induce specific antitumor cytotoxic T lymphocytes. Clin Cancer Res 2002; 8: 3885–92
- Lu J, Celis E. Recognition of prostate tumor cells by cytotoxic T lymphocytes specific for prostate-specific membrane antigen. Cancer Res 2002; 62: 5807–12
- Murphy G, Tjoa B, Ragde H, Kenny G, Boynton A. Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate 1996; 29: 371–80
- Tjoa B, Boynton A, Kenny G, Ragde H, Misrock SL, Murphy G. Presentation of prostate tumor antigens by dendritic cells stimulates T-cell proliferation and cytotoxicity. Prostate 1996; 28: 65–9
- Peshwa MV, Shi JD, Ruegg C, Laus R, van Schooten WC. Induction of prostate tumor-specific CD8+ cytotoxic T-lymphocytes in vitro using antigen-presenting cells pulsed with prostatic acid phosphatase peptide. Prostate 1998; 36: 129–38
- Inoue Y, Takaue Y, Takei M, Kato K, Kanai S, Harada Y, et al. Induction of tumor specific cytotoxic T lymphocytes in prostate cancer using prostatic acid phosphatase derived HLA-A2402 binding peptide. J Urol 2001; 166: 1508–13
- Dannull J, Diener PA, Prikler L, Furstenberger G, Cerny T, Schmid U, et al. Prostate stem cell antigen is a promising candidate for immunotherapy of advanced prostate cancer. Cancer Res 2000; 60: 5522–8
- Kiessling A, Schmitz M, Stevanovic S, Weigle B, Holig K, Fussel M, et al. Prostate stem cell antigen: Identification of immunogenic peptides and assessment of reactive CD8+ T cells in prostate cancer patients. Int J Cancer 2002; 102: 390–7
- Matsueda S, Kobayashi K, Nonaka Y, Noguchi M, Itoh K, Harada M. Identification of new prostate stem cell antigen-derived peptides immunogenic in HLA-A2(+) patients with hormone-refractory prostate cancer. Cancer Immunol Immunother 2004; 53: 479–89
- Friedman RS, Spies AG, Kalos M. Identification of naturally processed CD8 T cell epitopes from prostein, a prostate tissue-specific vaccine candidate. Eur J Immunol 2004; 34: 1091–101
- Kiessling A, Stevanovic S, Fussel S, Weigle B, Rieger MA, Temme A, et al. Identification of an HLA-A*0201-restricted T-cell epitope derived from the prostate cancer-associated protein prostein. Br J Cancer 2004; 90: 1034–40
- Kiessling A, Fussel S, Schmitz M, Stevanovic S, Meye A, Weigle B, et al. Identification of an HLA-A*0201-restricted T-cell epitope derived from the prostate cancer-associated protein trp-p8. Prostate 2003; 56: 270–9
- Oh S, Terabe M, Pendleton CD, Bhattacharyya A, Bera TK, Epel M, et al. Human CTLs to wild-type and enhanced epitopes of a novel prostate and breast tumor-associated protein, TARP, lyse human breast cancer cells. Cancer Res 2004; 64: 2610–8
- Carlsson B, Totterman TH, Essand M. Generation of cytotoxic T lymphocytes specific for the prostate and breast tissue antigen TARP. Prostate 2004; 61: 161–70
- Corman JM, Sercarz EE, Nanda NK. Recognition of prostate-specific antigenic peptide determinants by human CD4 and CD8 T cells. Clin Exp Immunol 1998; 114: 166–72
- Schroers R, Shen L, Rollins L, Xiao Z, Sonderstrup G, Slawin K, et al. Identification of MHC class II-restricted T-cell epitopes in prostate-specific membrane antigen. Clin Cancer Res 2003; 9: 3260–71
- Kobayashi H, Omiya R, Sodey B, Yanai M, Oikawa K, Sato K, et al. Identification of naturally processed helper T-cell epitopes from prostate-specific membrane antigen using peptide-based in vitro stimulation. Clin Cancer Res 2003; 9: 5386–93
- McNeel DG, Nguyen LD, Disis ML. Identification of T helper epitopes from prostatic acid phosphatase. Cancer Res 2001; 61: 5161–7
- Hural JA, Friedman RS, McNabb A, Steen SS, Henderson RA, Kalos M. Identification of naturally processed CD4 T cell epitopes from the prostate-specific antigen kallikrein 4 using peptide-based in vitro stimulation. J Immunol 2002; 169: 557–65
- Tjoa BA, Erickson SJ, Bowes VA, Ragde H, Kenny GM, Cobb OE, et al. Follow-up evaluation of prostate cancer patients infused with autologous dendritic cells pulsed with PSMA peptides. Prostate 1997; 32: 272–8
- Lodge PA, Jones LA, Bader RA, Murphy GP, Salgaller ML. Dendritic cell-based immunotherapy of prostate cancer: Immune monitoring of a phase II clinical trial. Cancer Res 2000; 60: 829–33
- Burch PA, Breen JK, Buckner JC, Gastineau DA, Kaur JA, Laus RL, et al. Priming tissue-specific cellular immunity in a phase I trial of autologous dendritic cells for prostate cancer. Clin Cancer Res 2000; 6: 2175–82
- Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, Valone FH. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 2000; 18: 3894–903
- Fong L, Brockstedt D, Benike C, Breen JK, Strang G, Ruegg CL, Engleman EG. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol 2001; 167: 7150–6
- Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest 2002; 109: 409–17
- Barrou B, Benoit G, Ouldkaci M, Cussenot O, Salcedo M, Agrawal S, et al. Vaccination of prostatectomized prostate cancer patients in biochemical relapse, with autologous dendritic cells pulsed with recombinant human PSA. Cancer Immunol Immunother 2004; 53: 453–60
- Burch PA, Croghan GA, Gastineau DA, Jones LA, Kaur JS, Kylstra JW, et al. Immunotherapy (APC8015, Provenge) targeting prostatic acid phosphatase can induce durable remission of metastatic androgen-independent prostate cancer: A Phase 2 trial. Prostate 2004; 60: 197–204
- Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity 1999; 10: 673–9
- Hernandez J, Garcia-Pons F, Lone YC, Firat H, Schmidt JD, Langlade-Demoyen P, Zanetti M. Identification of a human telomerase reverse transcriptase peptide of low affinity for HLA A2.1 that induces cytotoxic T lymphocytes and mediates lysis of tumor cells. Proc Natl Acad Sci USA 2002; 99: 12275–80
- Vonderheide RH, Anderson KS, Hahn WC, Butler MO, Schultze JL, Nadler LM. Characterization of HLA-A3-restricted cytotoxic T lymphocytes reactive against the widely expressed tumor antigen telomerase. Clin Cancer Res 2001; 7: 3343–8
- Arai J, Yasukawa M, Ohminami H, Kakimoto M, Hasegawa A, Fujita S. Identification of human telomerase reverse transcriptase-derived peptides that induce HLA-A24-restricted antileukemia cytotoxic T lymphocytes. Blood 2001; 97: 2903–7
- Schroers R, Huang XF, Hammer J, Zhang J, Chen SY. Identification of HLA DR7-restricted epitopes from human telomerase reverse transcriptase recognized by CD4+ T-helper cells. Cancer Res 2002; 62: 2600–5
- Schroers R, Shen L, Rollins L, Rooney CM, Slawin K, Sonderstrup G, et al. Human telomerase reverse transcriptase-specific T-helper responses induced by promiscuous major histocompatibility complex class II-restricted epitopes. Clin Cancer Res 2003; 9: 4743–55
- Vonderheide RH, Domchek SM, Schultze JL, George DJ, Hoar KM, Chen DY, et al. Vaccination of cancer patients against telomerase induces functional antitumor CD8+ T lymphocytes. Clin Cancer Res 2004; 10: 828–39
- Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4: 225–34