Abstract
To investigate the response of tumour growth to cisplatin treatment, in relation to p53 mutation and cyclin D1 dysregulation on DNA and protein level, biopsies from seven xenografted human squamous cell carcinomas from the head and neck were analysed with immunohistochemistry for p53 expression and cyclin D1 expression. Polymerase chain reaction -singlestranded conformation polymorphism was used to determine p53 mutations. Fluorescence in situ hybridization was performed to analyse cyclin D1 amplification. The mice were injected i.p. with NaCl (controls) or cisplatin. After injection the tumour volume were measured. The inhibition of tumour growth by cisplatin was defined as the area under the growth curves, and compared with the growth curves of the tumours in the control group. Xenografts with p53 mutation showed significantly higher resistance to cisplatin (p < 0.001) and also tumours with cyclin D1 amplification showed significantly higher resistance (p < 0.001).
Cisplatin is an alkylating chemotherapeutic drug used to treat several types of cancer, including squamous cell carcinoma of the head and neck (SCCHN). However, the response to cisplatin in SCCHN varies widely from total response to almost complete resistance. Cognetti et al. Citation[1] showed improved survival of patients with SCCHN who responded completely to cisplatin treatment. In a randomized study, Beauvillain et al. Citation[2] similarly demonstrated chemoresponse to be the most important prognosticator in patients with advanced laryngopharyngeal cancer. Better outcome with concomitant chemo and radiotherapy has recently been demonstrated Citation[3] while, neoadjuvant or adjuvant chemotherapy, combined with conventional therapy did not show improved survival Citation[4]. The effectiveness of treatment with cisplatin is restricted by intrinsic or acquired cellular resistance. The exact mechanisms governing cisplatin cellular uptake and inactivation have not been identified Citation[5]. The cellular response to cisplatin-DNA adducts, with respect to DNA repair, apoptosis, and/or cell cycle arrest, has been investigated yielding many contradictory results. One of the central genes in this context is p53. This gene is a transcription factor and activates downstream regulator genes in response to DNA damage i.e. p21 Citation[6]. We and others have shown a correlation between poor prognosis for patients with SCCHN and p53 mutation Citation[7], Citation[8]. A correlation has also been found between p53 mutation and cisplatin-fluorouracil neoadjuvant chemotherapy resistance Citation[9]. In contrast, Bradford et al. Citation[10] found that p53 mutation was correlated to cisplatin sensitivity in vitro. The cyclin D1 gene, CCND1, another cell cycle regulator, is frequently overexpressed and amplified in SCCHN. This is associated with poor prognosis Citation[11]. The aims of this study were to investigate the response of tumour growth to cisplatin treatment, in relation to p53 mutations and cyclin D1 dysregulation on DNA and protein level.
Methods and material
Tumours
To establish xenografted squamous carcinomas cell lines (SCC) biopsies were taken under general anaesthesia from patients with SCC not previously treated. The biopsies were then divided into two pieces, one for xenografting and one for in vitro cultivation. This study was approved by ethics committee.
The biopsies were xenografted to nude mice and serially passed as previously described Citation[16]. The cell lines studied originated from the following subsites: one from the larynx, one from the base of the tongue, four from the oral cavity and one from a lymph metastasis with the primary tumour unknown ().
Table I. Tumour origin and tumour line flow cytometry
To test the effect of chemotherapy, cell grafting was performed when the mice were 5 – 8 weeks old with a xenograft on each flank. Tumours were allowed to grow approximately two weeks by which time they had reached a sufficient volume and were in the growth phase.
Tumour volume and weight measurements
Three days before, and on the day of treatment the animals were weighed and the tumour volume measured, to ensure that no weight loss had occurred and that an appropriate tumour volume had been reached. Tumour volumes less than 40 mm3 and greater than 300 mm3 were excluded, as well as animals showing weight loss. The tumour volume was calculated from orthogonal diameter measurements every two or three days using the formula: volume = length×width2 /2 Citation[17]. The mice were weighed regularly during treatment
Treatment
On the day of cisplatin treatment, mice carrying the same tumour line were divided into four groups: the control group A was injected i.p. with NaCl; group B was injected i.p. with 2.5 mg/kg cisplatin body weight; group C 5.0 mg/kg and group D 7.5 mg/kg cisplatin body weight. The administered doses were diluted with saline to the required concentration and injected i.p. in volumes of 0.01 – 0.015 ml b.wt.
Cisplatin induced tumour growth inhibition
The tumour volume at day n relative to the tumour volume at the start of treatment (RTS) was transformed in toa 10log value in order to obtain a normal distribution. The area under the logRTS growth curve vs. time (AUC) was determined according to Lesser Citation[18]. The logRTS growth curves for the control group showed exponential growth and the doubling time of the tumour was determined. The AUC was calculated from the day of treatment to the day when the control group tumours had reached tripled in volume. A relative AUC value was calculated according to the formula: Relative AUC = AUC for the actual tumour /mean AUC value for the controls.
Immunohistochemistry
Untreated xenografts were allowed to grow for approximately two weeks, to reach sufficient size and to be in growth phase. Specimens were embedded in paraffin and immunohistochemical (IHC) analysis was performed to detect cyclin D1 and p53 expression. Commercial monoclonal antibodies were used (Novo, NCL-cyclin D1) at a dilution of 1:20 and p53 clone D0-7, Dako A-C Denmark, at a dilution of 1:300. A strongly positive specimen was used as a positive control. The IHC results () were scored as follows: A-negative; B 1 – 5% of the tumour cells positive; C 6 – 50% positive; D > 50% positive. The negative controls were tested without primer antibodies.
Fluorescence in situ hybridization
FISH, fluorescence in situ hybridization was performed as earlier described Citation[19] with minor modifications. Briefly, touch imprints from xenografted tumour cells were made on Superfrost Plus slides and then air-dried. These imprints were fixed in Carnoy′s solution (3:1 methanol:acetic acid) for 10 minutes and air-dried for one hour. The imprints were denatured in a solution of (70% formamide, 2xSSC, pH 7) at 72°C for three minutes and then dehydrated in a series of ethanol solutions (70%, 85% and 100%).
Two-colour hybridization was performed with a labelled gene-specific probe for CCND1 and with a labelled chromosome 11 centromere probe. A hybridization mixture containing the probe (LSI Cyclin D1 spectrum orange TM/CEP 11 spectrum green TM DNA Probe, 33-191039,vysis), 0.8 µl unmarked placenta DNA (D3287, Sigma) and 6.8 µl hybridization buffer (30-804826, vysis) was denatured in 72°C for 5 minutes and then hybridized at 37°C in a chamber overnight. After hybridization, the excess of the probe was washed away using (0.4xSSC, 0.3% Nonidet p40 at 72°C for 2 min; 2xSCC for 1min and then distilled water). DAPI (4,6-diamino-2-phenylindole) was added to a solution (Vectashield, Vector H1000) to visualize the DNA and to prevent the fluorochromates from fading. Zeiss fluorescence microscope was used to analyse the samples. The amplification rate was calculated as gene probe/centromere probe = copy number and was required to be over 1.5 to be considered as amplified. Tumour cell line no. 5 showed pronounced amplification and therefore marked with + + +.
PCR-SSCP and DNA sequencing
Polymerase chain reaction-single strand conformation polymorphism analysis (PCR-SSCP) and DNA sequencing, were used to study the occurrence of p53 mutations in exons 4 – 11, as previously described Citation[20]. Briefly, DNA was extracted using standard methods and used in a polymerase chain reaction to amplify the entire coding region of the p53 gene in seven or eight different fragments. The PCR products were screened for mutations by SSCP. Samples showing altered mobility shift in SSCP were further analysed by direct DNA sequencing to determine the exact location and type of mutation.
Flow cytometry (FCM)
FCM analysis was performed according to Wennerberg et al. Citation[16]. Briefly, the tumours were minced, forced through a nylon net (pore size 140 µm, Tidbeck AB, Stockholm, Sweden), and fixed in 70% ethanol. The separated cells were thereafter exposed to ribonuclease (Sigma-Aldrich, Stockholm, Sweden), incubated with pepsin solution (Merck, Darmstadt, Germany), and then stained with propidium iodide (Sigma-Aldrich, Stockholm). Human lymphocytes were processed in parallel with the tumour samples and used as an external diploid controls. Flow cytometric DNA analysis was performed in a FACS Caliber (Bacton, Dickinsen, Biosciences). DNA content was measured using an argon laser and up to 20 000 nuclei were analysed in each sample. The DNA histograms obtained were automatically processed using Modfit LT 3.1 software. The DNA indices (DI) were calculated as ratios of the modal channel values of G0/G1- peaks. By definition, the tumours manifesting a single DNA population were classified as diploid (with DI = 1.00), and tumours manifesting two or more populations as non-diploid. The s-phase fraction (SPF) was estimated assuming that s-phase compartment constituted a rectangular distribution between the modal values of G0/G1 and G2 peaks.
Statistics
Data were analysed with the RS/1 data analysis system. The normality of the distribution was tested with the Wilk-Skapiro test. Differences between groups were tested with one-way analysis (ANOVA) and the Kruskal-Wallis test.
Results
Expression of the p53 gene
As can be seen in , five out of seven tumour cell lines had p53 mutations; one of these had two different mutations. The mutations were located in exons 4, 5, 6 and 7. Three mutations were missense type (amino acid substitution), one nonsense, one a frame- shifting single base pair deletion and one a frame-three base pairs deletion. A previously described, a polymorphic variant (Arg72Pro) was also detected. Two xenografts had p53 mutations in introns + 72c > t and they were regarded as negative samples in our study. The three cell lines with missense or inframe-deletion mutations also showed p53 overexpression according to IHC, while the two cell lines with truncating mutations had no detectable protein expression. As expected, the two cell lines without p53 mutation had no p53 overexpression.
Table II. Description of p53 mutation, cyclin D1 dysregulation and relative AUC in xenografts. The immunohistochemical results were scored: A negative; B 1–5% of the tumour cells positive; C 6–50% positive; D > 50% positive.
Cyclin D1 expression
Four xenografted tumours showed CCND1 gene amplification, two of which also revealed overexpression of cyclin D1, as determined by IHC (). Three of the tumours showed no CCND1 amplification and no protein overexpression. Two tumour lines showed CCND1 amplification but no overexpression of the protein. All tumour lines with CCND1 amplification also had p53 mutation.
Cisplatin sensitivity
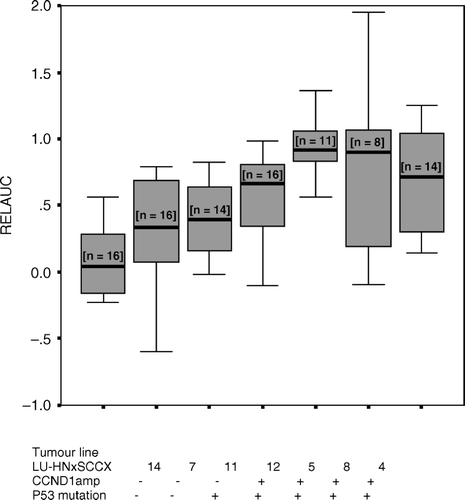
To compare the cisplatin sensitivity of the different tumour lines the dose of cisplatin 2.5 cisplatin mg/kg body weight was chosen. This low dose was chosen to better discriminate the differences in cisplatin sensitivity, since high doses effect both sensitive and resistant xenografts. The area under the growth curve was calculated and related to that of the controls (relative AUC). A higher relative AUC value indicates a higher resistance to cisplatin. A value of 1.0 indicates no growth inhibition as a result of treatment with cisplatin.
shows the sensitivity of the different tumour lines. The tumour lines LU-HNxSCC 4, 5, 8, 11, 12 with p53 mutations showed significantly lower cisplatin sensitivity than tumours with wt p53 (p < 0.001). Similarly, the tumour lines with CCND1 amplification showed significantly lower cisplatin sensitivity than those without amplification (p < 0.001). Cisplatin sensitivity was then compared between tumour lines with both p53 mutations and CCND1 amplification.The tumour line with only p53 mutations, showed significantly greater cisplatin sensitivity than that with both p53 mutations and CCND1 amplification (p = 0.004).
Figure 1. Boxplot diagram of relative AUC in various tumour lines, showing the 2½, 25, 50, 75 and 97½ cumulative relative frequencies. (n = number of tumour lines).
Flow cytometry
As can been seen in , all the tumour lines were non-diploid, with s-phase fraction of the tumour cells between 2.0 and 18.2%. There was no correlation between s-phase fraction and cisplatin sensitivity.
Discussion
Our results show that squamous cell carcinoma tumour lines from the head and neck without cyclin D1 gene amplification or p53 mutation respond better to cisplatin treatment (). DNA-sensing genes such as ATM, ATR or DNA-PK activate p53 by phosphorylation, by which p53 can induce cell cycle arrest or apoptosis. Missense or contact mutations of p53 have been related to poor prognosis Citation[8] and radioresistance in SCCHN in both clinical studies Citation[23] and in vitro models Citation[24]. Similarly, an increased sensitivity to cisplatin in patients with wtp53 has been reported Citation[9], although the converse has been demonstrated Citation[10]. P53 mutations have also been associated with increased radiosensitivity Citation[25]. The conflicting results could be due to differences in cell background, propensity for apoptosis and co-expression of oncogenes. Recently, additional proteins, such as p63 and p73, have been found to interact with p53 and to be involved in apoptosis and cell cycle regulation. A p53 polymorphism at codon 72, encoding either arginine (72R) or proline (72P), influenced cisplatin-based chemo-radiotherapy in advanced SCCHN, in that cancers expressing 72R mutants showed a lower response rate than 72P mutants. A possible explanation of this could be the involvement of p73-dependent apoptosis Citation[26]. A polymorphism has been noted in one tumour cell line, proline at codon 72, a tumour line that also had a nonsense mutation. We can not draw any conclusions regarding cisplatin response, from this finding.
The cyclin D1 gene/protein has been implicated in tumourogenes because of its importance in regulating G1 to S-phase checkpoint and consequently promoting cell cycle progression. Overexpression of cyclin D1 has been shown to stimulate cell proliferation Citation[27] and tumour progression Citation[28]. Thus, the use of antisense cyclin D1 in vitro in human squamous carcinoma, including head and neck, results in decreased cell proliferation, suppressed growth and the induction of apoptosis Citation[29]. Akervall and collaborators Citation[12] have reported that cyclin D1 overexpression indicated better response to neoadjuvant treatment with cisplatin and 5-fluoro-uracil. They have also shown that cyclin D1 overexpression in cell lines is associated with better response to cisplatin, however, no correlation was observed between cyclin D1 gene amplification and cisplatin sensitivity Citation[14]. In contrast, Ishiguro et al. Citation[13] have found an inverse relationship between cyclin D1 gene amplification and response to neoadjuvant treatment of SCCHN with cisplatin and 5-FU. A decrease in the expression of cyclin D1 results in a better response to cisplatin treatment in a variety of human cancers Citation[29] including SCCHN. Furthermore, studies have shown that antisense cyclin D1 enhances cisplatin sensitivity Citation[15]. The role of cyclin D1 gene amplification in cisplatin sensitivity could not be determined from our data in this in vivo setting, as none of the tumour lines showed only cyclin D1 gene amplification without p53 mutation.
Further investigations and more standardized methods are needed to find the key to DNA repair, cell arrest and apoptosis. Despite the small number of animals, our results imply there is a correlation between cisplatin response and the dysregulation of CCND1 and p53. Further studies are required with several xenografts and a more extended investigation of a combination of multiple genes.
This work was supported by the Foundations of the University Hospital of Lund, the Swedish Cancer Society (1304-B04-18XAC and 4839-B03-01PAC), the King Gustaf V Jubilee fund (04-4251) and governmental funding of clinical research within the NHS Region of Scania, R&D funding.
References
- Cognetti F, Pinnaro P, Ruggeri EM, Carlini P, Perrino A, Impiombata FA, et al. Prognostic factors for chemotherapy response and survival using combination chemotherapy as initial treatment of advanced head and neck squamous cell cancer. J Clin Oncol 1989; 7: 829–37
- Beauvillain C, Mahe M, Bourdin S, Peuvrel P, Bergerot P, Riviere A, et al. Final results of a randomized trial comparing chemotherapy plus radiotherapy with chemotherapy plus surgery plus radiotherapy in locally advanced resectable hypopharyngeal carcinomas. Laryngoscope 1997; 107: 648–53
- Pignon JP, Bourhis J, Domenge C, Designe L. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: Three meta-analyses of updated individual data. MACH-NC Collaborative Group. Meta-Analysis of Chemotherapy on Head and Neck Cancer. Lancet 2000; 356: 767–8
- Bourhis J, Pignon JP. Meta-analyses in head and neck squamous cell carcinoma. Hematology/Oncology clinics of North America 1999; 13: 769–75
- Kartalou M, Essigmann J. Mechanism of resistance to cisplatin. Mutation Research 2001; 478: 23–43
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31
- Koch WM, Brennan JA, Zahurak M, Goodman SN, Westra WH, Schwab D, et al. p53 mutation and locoregional treatment failure in head and neck squamous carcinoma. J Natl Cancer Inst 1996; 6(88)1580–6
- Mineta H, Borg A, Dictor M, Wahlberg P, Akervall J, Wennerberg J. p53 mutation, but not p53 overexpression, correlates with survival in head and neck squamous cell carcinoma. Br J Cancer 1998; 78: 1084–90
- Cabelguenne A, Blons H, de Waziers I, Carnot F, Houllier AM, Sousse, et al. P53 alterations predict tumour response to neoadjuvant chemotherapy in head and neck squamous cell carcinoma: A prospective series. J Clin Oncol 2000; 18: 1465–73
- Bradford CR, Zhu S, Ogawa H, Ogawa T., Ubell M, Narayan A, et al. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck 2003; 25: 654–61
- Åkervall J, Michalides R, Mineta H, Balm A, Borg Å, Dictor M, et al. Amplification of cyclin D1 in squamous cell carcinoma of head and neck and the prognostic value of chromosomal abnormalities and cyclin D1 overexpression. Cancer 1997; 79: 380–9
- Åkervall J, Michalides R, Dictor M, Brun E, Balm F, Wennerberg J. Cyclin D1 overexpression versus response to induction chemotherapy in squamous cell carcinoma of the head and neck–preliminary report. Acta Oncol 2001; 40: 505–11
- Ishiguro R, Fujii M, Yamashita T, Tashiro M, Tomita T, Ogawa K, et al. CCND1 amplification predicts sensitivity to chemotherapy and chemoradiotherapy in head and neck squamous cell carcinoma. Anticancer Res 2003; 23: 5213–20
- Akervall J, Kurnit DM, Adams M, Zhu S, Fischer SG, Bradford CR, et al. Overexpression of cyclin D1 correlates with sensitivity to cisplatin in squamous cell carcinoma cell lines of head and neck. Acta Otolaryngol 2004; 124: 851–7
- Marilene B, Wang MD, Helena T, Yip MD, Sriivatsan ES. Antisense cyclin D1 enhances sensitivity of head and neck cancer cells to cisplatin. Laryngoscope 2001; 111: 982–8
- Wennerberg J. Changes in growth pattern of human squamous cell carcinomas of head and neck during serial passages in nude mice. Int J Cancer 1984; 33: 245–50
- Steel G. Growth kinetics of tumours. Clarendon Press, Oxford 1977
- Lesser ML, Braun HI, Helson L. Statistical methods for measuring and comparing treatment efficacies: Applications to nude mice experimentation. Exp Cell Biol 1980; 48: 126–37
- Tanner MM, Karhu RA, Nupponen NN, Borg A, Baldetorp B, Pejovic T, et al. Genetic aberrations in hypodiploid breast cancer: Frequent loss of chromosome 4 and amplification of cyclin D1 oncogene. Am J Pathol 1998; 153: 191–9
- Fioretos T, Strömbeck B, Sandberg T, Johansson B, Billström R, Borg Å, et al. Isochromosome 17q in blast crisis of chronic myeloid leukemia and in other hematologic malignancies is the result of clustered breakpoints in 17p11 and is not associated with coding TP53 mutations. Blood 1999; 94: 225–32
- Manos MM, Ting J, Wright DK, Lewis AJ, Broker TR, Wollinsky SM. Use of polymerase chain reaction amplification for detection of human papillomaviruses. Cancer Cells 1989; 7: 209–14
- De Roda Husman AM, Walboomers JM, van den Brule AJ, Meijer CJ, Snijders PJ. The use of general primers GP5 and GP6 elongated at their 6′ends with adjacent highly conserved sequences improves human papillomavirus detection by PCR. J Gen Virol 1995; 76: 1057–62
- Raybaud-Diogene H, Fortin A, Morency R, Roy J, Monteil RA, Tetu B. Markers of radioresistance in squamous cell carcinomas of the head and neck: A clinicopathologic and immunohistochemical study. J Clin Oncol 1997; 15: 1030–8
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, et al. p53 status and the efficacy of cancer therapy in vivo. Science 1994; 266: 807–10
- Pekkola-Heino K, Servomaa K, Kiuru A, Grenman R. Increased radiosensitivity is associated with p53 mutations in cell lines derived from oral cavity carcinoma. Acta Otolaryngol 1996; 116: 341–4
- Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003; 3: 387–402
- Bartokova J, Lukas J, Muller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type Cyclin expression and G1 regulation in human head and neck cancer. Cancer Res 1995; 55: 949–56
- Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A 1994; 91: 709–13
- Sauter ER, Nesbit M, Litwin S, Klein-Szanto AJ, Cheffetz S, Herlyn M. Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res 1999; 59: 4876–81