Abstract
Cancer may arise because the developmental programs that create the dramatic alterations in form and structure in embryonic development are potentially corrupted. The cells in our bodies retain memories of these processes and cancer can occur later in life if imperfections occur in the fidelity of these pathways. This article is particularly interested in the phenomenon of epithelial to mesenchymal transition, which occurs in embryogenesis. Also reviewed are the small molecules and pathways that are involved both in homeostasis in adult epithelium and embryogenesis in utero. There are five such pathways in particular selected for review in this article: the Wnt pathway, Hedgehog, Notch, PAR and Bone morphogenetic peptide/TGF β. These are usually conserved throughout mammalian evolution. Though they have been arbitrarily separated in this article they are not exclusive from one another. Their pathologically altered expression is found especially frequently in childhood tumours where they may recapitulate their developmental role, and in tumours that resemble primitive precursor cells. These pathways are important for selecting cell fates, cellular rearrangements, cytological context and morphologic design in embryology as well as participating in epithelial function in adults.
The characterisation of the molecular biology of in-utero development and cancer has revealed remarkable similarities in their molecular origins. The common requirements of cellular proliferation and differentiation have considerable parallels in the signalling pathways that govern these processes. These include epithelial to mesenchymal transition, along with the Wnt, Hedgehog, Notch, PAR and BMP pathways, which are discussed in this review. Based on the accumulated knowledge derived from embryology and tumour biology, these pathways have been targeted for cancer therapy. This review does not cover every similarity but emphasises particular molecular patterns of potential future therapeutic importance.
Epithelial-mesenchymal transitions
In embryogenesis cells often have to alter their configuration and migrate to form a fetus. This is facilitated by epithelial-mesenchymal transition (EMT). EMT allows epithelial cells to change into mesenchymal cells, which have the ability to invade through extracellular matrix to create dramatic cell transpositions Citation[1]. It is decisive in gastrulation, when the ectodermal, mesodermal and endodermal layers are formed and in neural crest development. Cells undergoing EMT loose the apical-basal polarity of epithelial cells, down regulate cell adhesion molecule expression and induce the expression of vimentin filaments, fibronectin and N-cadherin (see ).
Figure 1. Diagram demonstrating the role of E-Cadherin; histologically, cellularly and molecularly in EMT. E-Cadherin, found at adherens junctions, maintains epithelial cell-cell contact, due to homophilic binding of its extracellular domains. During EMT, apical-basal polarity is lost, E-Cadherin expression is downregulated and mesenchymal genes are up-regulated. The binding of the transcriptional repressors Slug, Sip1, Snail and Twist to the inhibitory E-boxes of the E-Cadherin promoter terminates E-Cadherin expression.
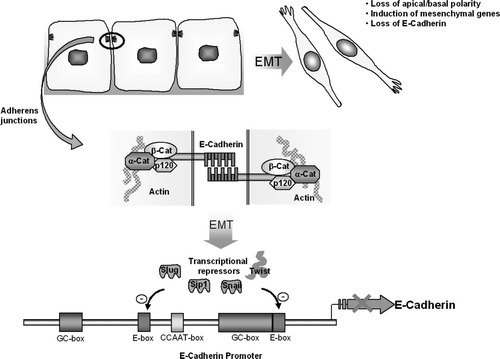
In EMT there is loss of E-cadherin, a homophilic calcium dependent cell adhesion molecule located in the adherents junctions of epithelia. Loss of E-cadherin expression is correlated with increased invasiveness of cancer cells. Reduced E-cadherin expression occurs when the zinc finger containing proteins Slug, Snail and SIP1 or the basic helix-loop-helix gene called twist down regulate the E-cadherin promoter by binding to a series of inhibitory E box sites (see ).
EMT in Embryogenesis
EMT has numerous roles in embryogenesis. This biological priority process is necessary for formation of the germ layers of the developing fetus and construction of organs and cellular rearrangements. provides an insight into some of the diverse impacts of EMT on fetal development as well as some instances of mesenchymal-epithelial transition (MET), (see ).
Table I. The importance of EMT and MET in embryogenesis. See text and associated cited references for additional information.
EMT in Cancer
EMT is implicated in the development of cancer especially gastric carcinoma. The Lauren classification classifies gastric carcinoma into either diffuse carcinomas made of discohesive cells that invade individually or intestinal gastric cancers composed of cohesive neoplastic cells that form glands and tubular structures. Snail expression has been found to inversely correlate with E-cadherin mRNA level in epithelial tumour cell lines of different origins. In one study Northern blotting established a reciprocal relationship between Snail and E-cadherin mRNA levels in most human epithelial cell lines examined. Furthermore transfection of MiaPaCa-2 cells with a human Snail antisense construction caused a significant decrease in Snail protein and a restoration of expression of E-cadherin mRNA and protein Citation[2]. In a study of 28 diffuse gastric carcinomas using real time quantitative RT-PCR reduced E-cadherin expression was detected in 11 (39%) and increased snail expression was found in six cases with reduced or absent E-cadherin expression Citation[3]. Germline truncating mutations in E-cadherin have been found in families with hereditary diffuse gastric cancer. Afflicted patients characteristically develop the cancer at an early age and pedigree analysis exhibits a highly penetrant autosomal dominant pattern of inheritance Citation[4].
The histological subtype of breast cancer is highly correlated with E-cadherin expression. E cadherin is usually expressed in invasive ductal carcinomas (IDC) and DCIS and its expression is usually lost in in-situ and invasive lobular carcinomas Citation[5]. Loss of E-cadherin expression causes the characteristic Indian file appearance of lobular breast carcinomas. In a landmark paper Yang et al. Citation[6] analyzed gene expression patterns associated with individual steps of mouse mammary tumour metastasis by utilizing a mouse mammary tumour model in which four distinct tumour cell lines reflected primary tumour growth, intravasation, extravasation and metastasis. This model facilitated selection of Twist as a candidate gene for modulating metastasis. To establish causality between Twist expression and tumour metastasis, highly metastatic 4T1 cells were transfected with lentiviruses transducing a DNA segment specifying short hairpin interfering RNA's (siRNA's) constructed against short coding regions of the mouse Twist gene. This dramatically reduced the expression of twist protein. Four weeks after implantation of 4T1 siRNA cells into mouse mammary glands examination of the lungs showed a reduced number of lung metastasis. Analysis of micro array gene expression from 57 human invasive breast tumours showed 70% of invasive lobular carcinomas exhibit increased twist expression compared to 32% of invasive ductal carcinomas. The interpretation of this research was that Twist is implicated in the intravasation step in metastasis in human lobular carcinomas Citation[6], Citation[7]. In infiltrating ductal carcinomas Snail has been found to be inversely correlated with tumour grade and in one study was expressed in all the IDC's presenting with lymph node metastasis Citation[8].
EMT may contribute to the development of neuroblastomas. These tumours arise from sympathetic neurons of neural crest origin and occasionally arise in-utero. There are at least three clinical patterns of disease: (i) spontaneously regressing widespread disease; (ii) loco-regional disease; and (iii) metastatic stage 4 disease with a 20% survival rate Citation[9]. Poor prognostic indicators for neuroblastmas include MYCN amplification, deletion of 1p, diploidy or tetraploidy and gain of chromosome arm 17q Citation[10]. N-myc oncogene amplification occurs frequently in neuroblastoma and is correlated with advanced disease stage and treatment failure. cDNA microarray analysis shows a link between Twist and N-Myc, as human twist is constantly overexpressed in N-myc amplified neuroblastomas. Twist inhibits the ARF/p53 pathway involved in the Myc dependent apoptotic response to cause dysregulated cellular proliferation and tumour progression Citation[11].
Furthermore, neuroblastoma cell culture investigations demonstrate spontaneous interconversion between neuroblastic cells and substrate adherent cells that resemble cultured glial or mesenchymal cells and have a molecular phenotype of cultured ectomesenchymal cells Citation[12]. This may be an analogous situation to the in vivo transition of malignant neuroblastomas into benign ganglioneuromas. Future research into molecular regulators of EMT may assist in explaining the apparently bizarre natural history of neuroblastomas with the tendency for stage 4S tumours to spontaneously regress.
WNT
The WNT signalling pathway
WNT's are cytosine rich glycoproteins that function as locally active molecules that bind to cellular receptors. There are nineteen members of the Wnt family in mammals alone and they are expressed in a spatially restricted and dynamic pattern in embryos and adults. If the local concentration of Wnt's exceeds the buffering capacity of the inhibitors they can interact with Frizzled receptors which are seven-transmembrane proteins and LRP co-receptors (LRP5/6 arrow) which are single transmembrane proteins that comprise a subfamily of LDL receptor related proteins. This interaction can activate the canonical β-catenin/TCF pathway and noncanonical pathways, including those mediated by Jun N-terminal kinase (Jnk) or calcium.
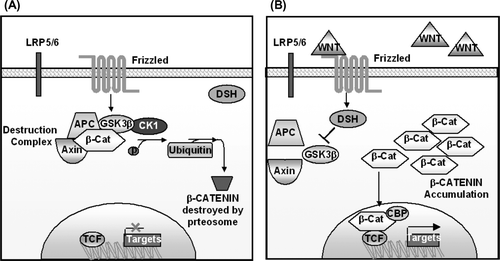
In the canonical/β-catenin pathway, in the absence of Wnt ligands beta catenin is recruited into a destruction complex. This contains the APC gene and AXIN, which facilitate the phosphorylation of β-catenin by casein kinase 1 and then glycogen synthase kinase 3. This leads to the ubiquitylation and proteosomal degradation of β-catenin (see a). Conversely when β-catenin levels are increased it is translocated to the nucleus where it binds to T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription elements (see b). Thus Wnt induces destabilisation of the beta-catenin destruction complex and stabilisation of β-catenin. This potentially affects the expression of downstream targets of wnt signaling including cyclins, c-myc and bone morphogenetic proteins.
Figure 2. Diagram of the Wnt signaling pathway. (A) In the absence of Wnt ligand, β-catenin undergoes intracellular destruction. (B) In the presence of Wnt ligand, β-catenin accumulates in the cytoplasm and translocates to the nucleus, where it binds to TCF/LEF transcription elements. This effects the transcription of downstream targets.
Wnt pathway and embryogenesis
In embryogenesis WNT's are involved in the induction of myogenesis Citation[13], tooth development Citation[14], CNS development especially of the telencephalon Citation[15] and early limb patterning Citation[16].
Wnt pathway and cancer
When errors occur in the destruction complex increased levels of β-catenin occur. A classical example is colonic carcinoma. Mutations in the APC gene are usually the first molecular alteration in sporadic colonic cancer and almost 100% of cases of sporadic colon cancer have deleted APC or mutated β-catenin which cause activation of the β-catenin pathway. β-catenin affects expression of cyclin D1 a cell cycle regulator, in colon carcinoma cells through TCF binding sites within the promoter Citation[17].
In familial adenomatous polyposis coli, germline mutations in the APC gene increase the chance of biallelic abnormal APC expression of a non-functional destruction complex and elevated β-catenin if a second somatic hit occurs according to Knudson's two hit hypothesis.
The effect of destabilisation of the destruction complex in development and cancer is provided by a familial case study. Wnt signaling genes are normally expressed in tooth development and tooth defects can be associated with defects in the members of the Wnt signaling pathway. A Finnish four-generation family in which there was dominant co-segregation of severe permanent tooth agenesis (oligodontia) and colorectal neoplasia was investigated with positional cloning to identify the causative mutation. A nonsense mutation Arg656Stop of AXIN2 was determined to be responsible Citation[18]. Additionally it has emerged that β-catenin mediated signaling is a promising strategy for early chemotherapeutic intervention. Data has suggested that sulindac, sulindac sulfone and indomethacin can modulate the subcellular localisation of β-catenin in vivoCitation[19].
Cyclin D1 overexpression had been found in approximately 50% of patients with breast cancer. In one paper examination of the promoter region of cyclin D1 found a perfect T cell factor (Tcf4) binding site. This implicates the β-catenin/TCF4 pathway in cyclin D1 expression in carcinoma of the breast. Transactivation of beta-catenin in eight breast cancer cell lines correlated significantly with cyclin D1 expression. Furthermore in 123 patient breast tumour samples, immunohistochemistry showed beta-catenin and cyclin D1 expression were significantly correlated. Activation of β-catenin and cyclin D1 over expression was established as associated with poorer prognosis on Kaplan-Meier analysis and the log rank test Citation[20].
Alterations of the Wnt pathway are associated with hepatocellular carcinoma Citation[21], and anaplastic carcinoma of the thyroid gland Citation[22]. β-catenin mutations occur in medulloblastomas, hepatoblastomas and Wilm's tumours Citation[23]. Wilm's tumours can occur in Denys-Drash syndrome, a familial disorder secondary to germline mutations in WT1 Citation[24]. Wnt 1 functions as a mammary oncogene in mouse mammary tumour virus infected mice Citation[25]. In intestinal type gastric carcinomas 27% harbour β-catenin mutations Citation[26]. Desmoid tumours have increased levels of beta-catenin usually due to mutations in APC or point mutations in beta-catenin Citation[27]. β-catenin also functions in hair follicle morphogenesis and β-catenin gene mutations affecting exon 3 have been implicated in human pilomatrixomas by activating gene expression together with lymphoid enhancer factor 1 Citation[28].
Hedgehog/Gli
The hedgehog/Gli pathway
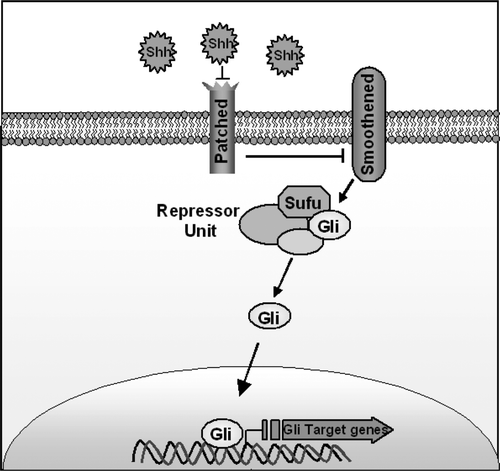
There are three glycopeptide hedgehog family members in mammals, Sonic (Shh), Indian (Ihh) and Desert (Dhh). Signalling in the sonic hedgehog pathway pathway is affected by the secreted hedgehog ligands binding to a membrane receptor complex called patched (Ptch). Ultimately this hedgehog-patched (Hh-ptch) interaction activates GLI zinc finger transcription factors. Two intermediates in this pathway are the cell receptor smoothened (SMOH) and the cytoplasmic molecule SUFU. The Hh-ptch interaction relieves inhibition on smoothened (SMOH), a seven transmembrane receptor that up regulates PTCH itself and Gli zinc finger transcriptional factors. SUFU forms a repressor unit that inhibits Gli activity (see ).
Figure 3. Diagram of the Sonic Hedgehog pathway. Sonic Hedgehog binds to the Ptch receptor, this relieves inhibition of Smoothened. Downstream this causes upregulation of Gli zinc finger transcription factors. SUFU is an intracellular molecule that forms a repressor unit that inhibits Gli activities.
Gorlin's syndrome, SUFU mutations and exogenous poisons
Aberrations may affect different steps in the Shh Hedgehog/Gli pathway. Gorlin's syndrome an autosomal dominant condition arises by germline mutations in the PTCH gene and is characterised by developmental abnormalities, multiple basal cell cancers, medulloblastoma-usually the desmoplastic subtype and meningiomas. In a series of 46 tumour samples from patients with medulloblastomas, sequence analysis and RT-PCR found truncating mutations of SUFU in 9%, which correlated with the desmoplastic variant. Missense mutations of SUFU; P15L and A340S, also occurred. The P15L mutation and truncating mutations were accompanied by LOH, loss of the wild type allele or both Citation[29]. Environmental toxins can target another signalling component of the hedgehog/Gli pathway, Smoothened (SMOH). Exposure to the teratogenic alkaloid cyclopamine a hedgehog pathway antagonist in the plant Veratrum californicum has been found to cause a congenital holoprosencephalopathy in studies on livestock Citation[30], Citation[31]. This arises through inhibition of the Shh signalling pathway at the level of the smoothened receptor (SMOH).
Hedgehog and embryogenesis
The hedgehog glycoproteins sonic, Indian and Desert have important roles in embryonic development (see ).
Table II. The role of Sonic Hedgehog, Desert Hedgehog, Indian Hedgehog and Ptch in embryogenesis. See text and associated cited references for additional information.
Hedgehog and cancer
Aberrations in the Hedgehog signalling pathway can lead to cancer. This may be secondary to sustained increased endogenous expression of Hh ligands or ligand independent mutational activation of the Hh pathway. Increased SHH and IHH mRNA has been detected in 97% of cell lines (37/38), including oesophageal, stomach, biliary tract and pancreatic tumours, but not found in (0/11) colonic tumour cell lines Citation[32]. Assessment of hedgehog pathway activation by RT-PCR in tumours growing in vivo determined PTCH mRNA levels to be 23–271 times higher in stomach tumours and 69-5044 times higher in pancreatic tumours than in adjacent normal tissue Citation[32].
Tumours involving activation of the Hedgehog pathway may also arise due to mutation of pathway components. A classical example is medulloblastomas in particular the desmoplastic variant. These tumours may arise through inappropriate constitutive activation of the ShGli pathway although they may arise in other ways such as by abnormalities in the tumour suppressor genes retinoblastoma and p53 Citation[33]. Medulloblastomas are the most common malignant CNS tumour of childhood and have a peak incidence in children between five and nine years of age. They arise from immature granule cells of the cerebellum. Maintenance of abnormal progenitor-like fates through Hh pathway activation causes malignant growth of these tumours. Loss or mutation of the PTCH allele occurs in 10–20% of cases of medulloblastoma. With respect to SUFU, fluorescent in situ hybridisation (FISH) studies have mapped SUFU to chromosome 10q24.3, loss of chromosome 10q occurs in 41% of cases and mutations of SUFU has been described in individual cases Citation[29], Citation[34]. In normal tissues Gli proteins are especially expressed in precursor cells. It is conceptually possible that the tumours arising from increased Hh-Gli activation are recapitulating primitive developmental biology or they arise from stem cells that cannot differentiate or halt proliferation. Sonic hedgehog (Shh) is expressed in Purkinje neurons on the normal cerebellar cortex and in development Shh usually promotes proliferation of granule neuron precursors of the external germinal layer of the cerebellum Citation[35].
Alterations in Hedgehog signalling also occur in other malignancies. Shh signalling is required for branching morphogenesis in lung development and Shh and Gli 1 expression are increased in airway epithelium regeneration post acute lung injury just prior to neuroendocrine differentiation. Experimental evidence from a murine model examining Clara cells (a specialised distal airway epithelial cell) 72 hours post systemic administration of naphthalene demonstrated increased expression of both SHH ligand and Gli1. Separate analysis of ten small cell lung cancers (SCLC) showed 5/10 exhibited increased Shh and Gli1, and 7/7 SCLC cell lines demonstrated increased Shh protein expression. Similarly to hedgehog pathway activation in digestive tract tumours, increased hedgehog pathway activation in SCLC may arise from pathway activation secondary to regeneration post tissue injury and both groups of tumours arise from tissues of endodermal origin Citation[36]. In cutaneous malignancies single strand conformational polymorphism analysis followed by DNA sequencing of 42 sporadic basal cell carcinomas (BCCs) demonstrated PTCH mutations in 67% (28/42), SMOH mutations in 10% (4/42) and mutations in SUFU in two cases. Microsatellite markers also revealed LOH on 9q22, the chromosomal location of PTCH, in 53% of cases. Thus, there was at least one PTCH alteration in 83% of BCCs assessed, with biallelic inactivation of PTCH in 42% of cases Citation[37].
The pathways described are not exclusive. An instance of interaction between two of the pathways described thus far is provided by Turcot's Syndrome (colon cancer and medulloblastomas). A subset of children with medulloblastomas has germline mutations of the SUFU gene. In murine models SUFU can bind β-catenin and decrease β-catenin/Tcf transcription. Potentially loss of function of SUFU can result in over activity of the Sonic Hedgehog and Wnt signalling pathways leading to dysregulated cellular proliferation Citation[38].
Notch
The notch-signalling pathway
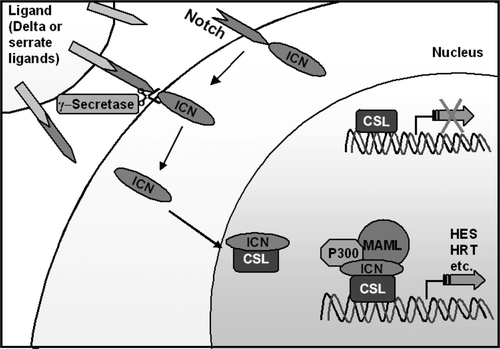
Notch is a highly conserved evolutionary pathway that controls cell fate specification, stem cell maintenance and initiation of differentiation in embryonic and postnatal tissues Citation[39]. It's name arose because when the first notch receptor was described, haplo-insufficiency of the gene was found to cause notches on the wings of flies. The effects of the notch-signalling pathway depend on the cellular context in which it acts to regulate cell proliferation, differentiation and binary cell fate decisions. In mammals four notch genes (NOTCH1-4) are expressed, cleaved and incorporated onto the cell surface as a heterodimers. Their extra cellular domains contain between 29 and 36 epidermal growth factor like repeats that bind ligands on the extra cellular surface of neighbouring cells Citation[40]. The Notch heterodimer has a single transmembrane domain and its cytoplasmic extension conveys the signal to the nucleus. The pathway is activated by delta (delta like ligands 1, 3, or 4) or serrate ligands (jagged 1 or 2). When notch is stimulated by its ligands on adjacent cells the intracellular domain of notch (Notch-IC) is cleaved by metalloprotease and γ-secretase. This causes nuclear translocation of notch-IC (see ). Notch-1C binds to CSL and converts it from a transcriptional repressor to an activator by displacing inhibitory proteins and recruiting co-activators including Mastermind and histone acetyltransferases. This multiprotein enhancesome results in the transcription of target genes of the notch signalling pathway. Downstream target genes are still being characterised, classical targets are the Hairy/ Enhancer of Split (HES) and hairy related families Citation[41]. Notch signalling is regulated by endocytosis, glycosylation and ubiquitination. The notch ligand intercellular interaction can be lateralising or inductive. This means that depending on the stochiometric relationships between ligands and notch receptors intra- and intercellularly, activating the pathway results in distinct cellular context mediated cell fates. Frequently the net result of notch signalling is to preserve a small population of uncommitted multipotential cells within a differentiating tissue Citation[42], Citation[43]. Ultimately the notch pathway is responsible for stem cell maintenance, binary cell fate decisions and induction of differentiation.
Figure 4. Diagram of the Notch Pathway: Pathway activation by delta or serrate ligands cleaves the Notch receptor and causes translocation of its intracellular portion (ICN) to the nucleus. ICN forms part of an enhancesome that results in the transcription of target genes.
Aberrant notch expression in malignancy
Notch can function as an oncogene or tumour suppressor gene depending on the cellular context and the interaction with other signal-transduction pathways (see ). Aberrations in notch expression have been implicated in solid and haematological malignancies. Cancer may arise because of constitutive activation of the NOTCH receptor, ligand independent activation of its transcriptional target genes or alterations in the levels of delta or serrate ligands.
Table III. Alterations in the Notch Pathway in selected malignancies
The best characterised role of Notch signalling in haematopoiesis and lymphopoiesis is the essential role of Notch1 in T-cell lineage commitment Citation[44]. This role may be corrupted as the chromosomal translocation t (7; 9) (q34; q34.3) in human T cell acute lymphoblastic leukaemia (T-ALL) results in the constitutive activation of notch. This chromosomal translocation occurs in less than 1% of T-ALLs, however, more than 50% of human T-ALLs have activating mutations that involve the extra cellular heterodimerization domain and/or the C-terminal PEST domain of Notch1 Citation[45].
Notch also causes solid malignancies including mucoepidermoid carcinoma the most common malignant salivary gland tumour Citation[46]. A t (11; 19) (q14-21; p12-13) translocation frequently occurs in mucoepidermoid carcinoma and occasionally is the sole cytogenetic change. This translocation fuses exon 1 from a gene mucoepidermoid carcinoma translocated 1 (MECT 1) with exons 2–5 of a novel member of the Mastermind-like gene family (MAML2) at 11q21. Spectral karyotyping of two epidermoid carcinoma tumour cell lines identified this translocation, and RT-PCR of RNA extracted from these cell lines and tumour biopsy samples confirmed expression of the chimeric product. A tumourigenic assay found that MECT-1-MAML2 induced foci of RK3E cells, which are epithelial cells immortalized with adenovirus E1A (RK3E cells). In vitro tests confirmed activation of the HES1 promoter and expression of HES1 by the MECT-1-MAML2 fusion product. Activation of the target occurs independently of notch ligand and CSL binding sites Citation[44].
Altered notch signalling occurs in tumours of the central nervous system that have a childhood predisposition such as neuroblastomas and medulloblastomas. This aberrant notch signalling causes failure of terminal neuronal differentiation, a permanent dedifferentiated state and persistent expression of notch target genes that are normally or predominantly expressed in embryogenesis. This is a classical example of the maturation arrest theory of pathogenesis applied to specific cancers. For example in the majority of neuroblastomas mammalian achaete-scute homologue 1 (MASH-1 or HASH-1 in humans) is expressed, this is normally only transiently expressed in migrating sympatho-adrenal cells indicating the tumour recapitulates corrupted embryonic development. HASH-1 is negatively controlled by HES-1 a transcriptional target of the notch signalling cascade Citation[47]. When neuroblastoma cells are induced to differentiate HASH-1 expression is rapidly downregulated with concomitant, transient upregulation of HES-1 Citation[48]. Potentially neuroblastoma development may be related to abnormal temporal expression of the HASH-1 and HES-1 genes. Similarly in medulloblastomas Notch2 receptor transcripts are highly expressed with minimal or no Notch1. This parallels normal cerebellar development in which notch 2 is predominantly expressed in proliferating progenitors and notch1 in postmitotic differentiated cells Citation[49].
Lung cancer ontology may arise through dysregulated pleuripotent stem cells. In a study of two small cell lung cancer cell lines, notch induced a G1 cell cycle arrest associated with p21waf/cip1 induction, repression of Hash1, and induction of the downstream ras signalling pathway. Citation[50]. In other cancers Notch1 functions as a tumour suppressor in mouse skin Citation[51] and interacts with HV16 E6 and E7 oncoproteins in cervical cancer Citation[52].
Quantitative abnormalities in the levels of notch ligands also have been implicated in cancer development. In a study of 154 men with prostate cancer immunohistochemistry for JAGGED 1 expression found it to be more highly expressed in metastatic as compared with localised prostate cancer. High JAGGED 1 expression also was correlated with recurrence of localised prostate cancer independent of other clinical parameters. This suggests that Jagged1 expression may be used to distinguish indolent and aggressive prostate cancers Citation[53]. In a study of JAG2 expression in patients with multiple myeloma and 4 multiple myeloma cell lines, JAG2 expression was increased compared to controls. Its expression was also increased in (5/5) patients with monoclonal gammopathy of uncertain significance. This suggests that JAG2 over expression is an early event in the development of multiple myeloma. Anti-notch-1 monoclonal antibodies directed against its binding sequence with JAG2 blocked the in-vitro secretion of IL-6. Citation[54].
Protease activated receptors (PAR's)
Activation of PAR's
In addition to facilitating the spread of cancers through degradation of extracellular matrix proteins, proteases participate in cancer biology through a family of proteinase-activated receptors (PAR's). These are a family of serpentine G protein coupled receptors that occur in organisms from C. elegans to humans. Four members of the PAR's are known. PAR-1, PAR-3 and PAR-4 are activated by the serine protease thrombin and PAR-2 is activated by trypsin like enzymes. PAR's are widely distributed throughout the body and play a role in embryogenesis, physiology and disease. Their activation does not occur through ligand binding; instead activation is mediated by cleavage of the N terminus of the receptor by a serine protease generating a tethered ligand that interacts with the receptor within extracellular loop 2. PAR receptors have been implicated in the metastatic phenotype, may interact with cellular integrins, and intracellularly may phosphorylate the MAP kinase pathway in some cancers. Thrombin is a serine protease that activates PAR's has a crucial role in the coagulation cascade and is implicated in angiogenesis, tumour cell-endothelial cell adhesion, and invasion. Increased coagulation factors occur in many cancers both systemically and participate in autocrine loops locally. Thrombin antagonists are a potential treatment for malignancies.
PAR's in embryogenesis
Protease activated receptors participate in embryogenesis and in development of the oocyte. Anterior posterior asymmetry of microtubules in C. elegans embryos depends on PAR-3 protein and heterotrimeric G protein signalling. PAR-2 affects microtubule dynamics by restricting PAR-3 activity to the anterior embryo Citation[55]. In early development determinants of future embryonic axis become asymmetrically distributed within the oocyte. PAR-1 functions in anterior posterior cell patterning, and it appears that Drosophila PAR-1 functions by phosphorylating proteins that regulate microtubule dynamics. In particular it phosphorylates a protein called Osk that patterns the posterior body plan in Drosophilia. PAR-3, a PDZ scaffold protein participates in cell membrane junction formation Citation[56]. The zebrafish PAR-3 ortholog (pard3) is restricted to the outer limiting membrane and outer and inner plexiform layers in the retina after retinal lamination. Reducing pard3 expression results in cell death in the ventral diencephalon and cyclopia with failure of separation of the eye fields Citation[57].
PAR-1 in cancer
The development of cancer particularly when metastatic has been associated with induction of the PAR-1 gene. High PAR-1 expression has been reported in laryngeal carcinoma Citation[58], renal carcinoma Citation[59], breast carcinoma Citation[60], lung adenocarcinoma Citation[61] and oral SCC Citation[62]. Sustained constitutive activity of PAR-1 occurs in metastatic breast cancer cell lines because of altered trafficking of the activated PAR-1 receptor secondary to decreased lysosomal degradation Citation[63]. The metastatic ability of breast cancer is correlated with PAR-1 expression levels and PAR-1 mediated breast tumour cell invasion involves co-operation with ανβ5 integrin Citation[64]. This has a physiological correlate as PAR-1 is temporally expressed in the cytotrophoblast layer between 7 and 10 weeks gestation. Furthermore matrix metalloproteinase-1 in the stromal tumour microenvironment functions as a PAR-1 agonist to promote invasion and tumourigenesis of breast cancer cells Citation[60].
In colon cancer cells PAR-1 is ectopically expressed, it is absent in normal colonic mucosa. Aberrant expression of the PAR-1 receptor increases cell proliferation and motility in colonic carcinoma. Colonic cancer involves systemic activation of the coagulation system and generation of thrombin a serine protease that can activate protease-activated receptors. Increased thrombin/antithrombin complexes have been found in particular in the venous drainage of patients with colonic tumours Citation[65]. Thrombin induces phosphorylation of the protein kinases p42/p44 and thrombin activation of MAP kinases participates in the thrombin mediated events in colon cancer Citation[66]. Therefore colonic cancer can progress by thrombin generation and consequential PAR-1 cleavage.
The metastatic potential of human melanoma cells has also been found to correlate with PAR-1 over-expression Citation[67]. It is to be noted that PAR-1 expression is not invariably associated with increased metastatic ability however. In squamous cell carcinoma of the head and neck PAR-1 is required for the expression of involucrin, a keratinocyte differentiation marker, and it's expression is inversely correlated with cervical lymph node metastasis Citation[68].
PAR-2 in cancer
Trypsin a ligand of PAR-2 has been implicated in many cancers including digestive tract tumours. It participates in autocrine/paracrine loops in colonic tumours. In colon cancer it induces growth and invasion by transactivation of the epidermal growth factor receptor (EGF-R) by PAR-2. This occurs by TGF-α mediated activation of the EGF-R and consequential EGF-R phosphorylation and activation of ERK1/2 Citation[69].
Protease activating receptor-2 is frequently expressed in papillary adenocarcinoma of the gallbladder. Pancreaticobiliary maljunction with reciprocal reflux of bile and pancreatic juice is a risk factor for gallbladder carcinoma and theoretically trypsin mediated PAR-2 stimulation may have an important role for carcinogenesis of the gallbladder Citation[70]. In human gastric carcinoma cells trypsin activation of PAR-2 stimulates integrin α5β1–dependent adhesion to fibronectin and proliferation Citation[71]. PAR-2 activation by trypsin also plays an important role in promoting proliferation of pancreatic cancer Citation[72].
Thromboembolic events occur with increased frequency in patients afflicted with cancer and is patients with GBM's particularly Citation[73]. Human gliomas express prothrombin Citation[74] and meizothrombin an intermediate of prothrombin activation stimulates glioblastoma cells by interaction with PAR-1 type thrombin receptors Citation[75]. Glioblastoma cell lines have been found to express PAR-1 and 2 Citation[76]. Potentially GBM's may progress by an autocrine mechanism of increased serum thrombin levels, which activate PAR's on the tumour cells surface. In addition to mitogenesis thrombin enhances vascular endothelial growth factor induced endothelial cell proliferation Citation[77]. Vascular endothelial hyperplasia is a characteristic feature of GBM's.
Protease activated receptors also participate in various other miscellaneous cancers. A membrane bound serine protease TMPRSS2 has localised expression in the prostate, activation releases it to the extracellular space with consequent activation of PAR-2 in prostate cancer cells Citation[78]. This may contribute to prostate cancer metastasis. Activation of PAR-1 or PAR-2 in prostate cancer cell lines increase levels of the proteolytic enzymes MMP-2 and MMP-9 implicating PAR receptors in an increased tendency for metastasis Citation[79].
PAR-4 and cancer
PAR 4 is a pro-apoptotic protein and its downregulation has been linked to many cancers. It is required for neuronal apoptosis and has been shown to experience down regulation in several CNS tumours Citation[80].
Bone morphogenetic peptides
The BMP pathway
Transforming growth factor-β members include TGF-β, activins and bone morphogenetic proteins. These are structurally related cytokines secreted in diverse Metazoans Citation[81]. In most circumstances, expression of TGF-β pathways has a tumour suppressor effect. Discussion of TGF-β in it's entirely is beyond the scope of this review, therefore particular emphasis is placed on bone morphogenetic peptides in selected malignancies. Thirty different BMP's in invertebrates and vertebrates have been identified. BMP's are extracellular ligands that bind to serine/threonine receptors and phosphorylate intracellular Smad proteins. Smads serve as transcriptional regulators, specifically Smad 1, 5 and 8. These phosphorylated Smad's homo and heterodimersise with Smad 4 a common partner. This complex enters the nucleus subsequently to alter target gene expression Citation[82]. Three classes of Smad receptors exist, (a) Receptor regulated Smads (R-Smads), Smads 1,2,3 and 5 (b) Common Smads (Co Smads), Smad4 which forms heteromeric complexes with the R-Smads and (c) Inhibitory Smads (I-Smads), Smad6 and Smad7 Citation[83].
BMP's in embryogenesis
In embryogenesis BMP's affect left-right asymmetry, distal limb morphogenesis, mesoderm formation in gastrulation, induction of endochondral bone formation by BMP-2 cell migration Citation[84], differentiation and apoptosis. TGF-β signals including BMP4 and BMP7 are required for morphogenesis of the kidney mesenchyme progenitor population Citation[85]. BMP's regulate patterning of embryonic tracheal epithelium Citation[86], patterning of the cerebral cortex Citation[15] and also have been implicated in vasculogenesis in embryos.
BMP's in cancer
In cancer, BMP's have been implicated in lymphomas, colonic cancer, lung tumours and skeletal metastasis especially. Overexpression of Smad 1 occurs in follicular lymphoma cells and is characterised by the t (14; 18) bcl2/IgH translocation Citation[87]. In adult T cell leukaemia and tropical spastic paraparesis, human T cell lymphotropic virus type 1 is a causative factor. Tax a 40kDa protein, which is encoded by HTLV-1 pro-viral DNA, inhibits transcriptional activation and growth inhibitory responses to TGF-β. This occurs in part by Tax disrupting the association of Smads with the transcriptional co-activator p300, direct interaction with Smad2, 3 and 4 as well as inhibition of Smad3: Smad4 complex formation and it's binding with DNA Citation[88]. It has also been found that bone morphogenetic proteins 2, 5, 6 and 7 inhibit growth and induce apoptosis in myeloma cells Citation[89]. Potentially BMP analogues may offer a promising therapeutic strategy in myeloma.
Aberrations in TGF-β/BMP have been implicated in gastrointestinal malignancies. Frequent Smad4 mutations have been found in colon cancer. Smad4 is located at chromosomal region 18q21 a site of LOH in colorectal cancer and patients with a normal Smad4 diploidy have a three fold higher benefit of 5-fluorouracil based adjuvant chemotherapy Citation[90]. In colonic cancer, BMP-2 may act as a tumour suppressor gene in mature colonic epithelial cells, as Bmp2 inhibits colonic epithelial cell growth in vitro and BMP2 expression is lost in adenomas of patients with familial adenomatous polyposis coli Citation[91]. Bmpr Ia and Smad4 mutations have also been found in some cases of juvenile polyposis Citation[92].
BMP's are capable of promoting the differentiation of mesenchymal cells into osteogenic lineage pathways and frequently have a critical role in the epithelial stromal interaction in the microenvironment of bone metastasis. Bone provides chemotactic factors, adhesion factors and growth factors that allow prostate carcinoma cells to target and proliferate in the skeleton Citation[93]. In prostate cancer bone morphogenetic proteins and endothelin 1 may stimulate differentiation of osteoblast precursors to mineral producing osteoblasts. It has been found that in prostate cancer metastasis BMP's in particular BMP-7 induce VEGF protein expression through activation of the promoter 4 Citation[94] and VEGF promotes osteoblastic metastatic lesions in prostate cancer. Elevated levels of BMP-6 correlates with skeletal metastases in prostate cancer Citation[95], recombinant noggin (a BMP antagonist) inhibits the function of BMP-6 and indicates a possible therapeutic strategy for painful osteosclerotic bone metastasis in prostate cancer.
Problems with correlative evidence
In this paper a retrospective literature review was performed on selected genes of potential relevance for particular types of cancer. Correlative evidence with papers in developmental biology has usefulness in demonstrating the biological diversity of the maturation arrest theory of cancer biology in specific malignancies. Mining the available literature for correlative evidence between molecular aberrations, human tumours and the physiological relevance of individual genes in embryology poses specific technical difficulties. By necessity this approach is retrospective, not uniform in molecular techniques, frequently depends on in-vitro data and has to accommodate evolutionary heterogeneity when studying tissue from other species. A retrospective literature review is valuable as it consolidates current information, allows an intuitive appraisal of emerging patterns of evidence and guides selection of further molecular studies that include the experimental rigour required to demonstrate pathogenic significance of molecular changes. Of relevance to this problem bioinformatics techniques allow selection of common critical genes in embryology and cancer in an experimentally prospective way. Furthermore the Gene Ontology Consortium has provided a freely available computerised comparison between differing databases; of molecular function, biological process and cellular components using structures called directed acyclic graphs. Also a consensus of human cancer genes has been established allowing determination of structural and functional properties of genes involved in cancer Citation[96], Citation[97].
High throughput techniques that assess DNA, RNA and protein can determine genes of potential relevance. Modern genome-wide screening tools, such as cDNA microarrays Citation[98], and GeneChip arrays (oligo arrays) Citation[99] has revolutionized cancer research. Microarrays have allowed the study of thousands of genes simultaneously providing a comprehensive view of the genetic mutations and their consequences. The fundamentals of microarray technology can also be adapted to suit genomic DNA analysis, such as DNA methylation analysis, mutational analysis, polymorphism genotyping and comparative genome hybridization (CGH) Citation[100]. Likewise, discovery based techniques for proteomics include two dimensional gel electrophoresis (2-DE gels), mass spectrometry and chromatography.
These techniques along with bioinformatics tools allow selection of genes of potential significance. Correlating gene abnormality to cancer development requires functional experiments. Cancer is considered to arise from alterations in six essential regulatory systems called the hallmarks of cancer: insensitivity to growth inhibitory signals, evasion of apoptosis, self sufficiency in growth signals, limitless replicative potential, sustained angiogenesis and tissue invasion and metastasis Citation[101]. In this review we attempted to correlate molecular aberration with these alterations and the malignant phenotype.
Conclusion
Death from cancer is as much a part of life as birth due to development. The striking similarity in the pathways involved in embryogenesis and cancer, the potential for corruption of the balance between life and death and the theoretical therapeutic targets are all being unravelled. Exciting therapeutic opportunities exist. For instance, small intracellular molecules called pepducins that affect the transmission of signals from activated protease activated receptors to their associated G proteins may be useful in inhibiting metastasis Citation[102]. An investigational new drug application has been filed with the FDA in 2005 for a topical hedgehog antagonist for basal cell carcinoma. A phase I clinical trial of a notch signalling pathway inhibitor for patients with T-cell acute lymphoblastic leukaemia/lymphoma is currently recruiting patients (ClinicalTrials.gov, Identifier: NCT00100152). Numerous other therapeutic developments may arise from elucidating these pathways. New techniques such as forward genetics are identifying genes involved in embryology with implications for cancer research. It is difficult to be certain what will be the therapeutic consequences of this accumulated knowledge, but certainly the end and beginning of existence has more in common than we have previously realised.
References
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995; 154(1)8–20
- Batlle E, Sancho E, Franci C, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2(2)84–9
- Rosivatz E, Becker I, Specht K, Fricke E, Luber B, Busch R, et al. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol 2002; 161(5)1881–91
- Huntsman DG, Carneiro F, Lewis FR, MacLeod PM, Hayashi A, Monaghan KG, et al. Early gastric cancer in young, asymptomatic carriers of germ-line E-cadherin mutations. N Engl J Med 2001; 344(25)1904–9
- Acs G, Lawton TJ, Rebbeck TR, LiVolsi VA, Zhang PJ. Differential expression of E-cadherin in lobular and ductal neoplasms of the breast and its biologic and diagnostic implications. Am J Clin Pathol 2001; 115(1)85–98
- Yang J, Mani SA, Donaher JL, Ramasamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117(7)927–39
- Kang Y, Massague J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004; 118(3)277–9
- Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002; 21(20)3241–6
- Mora J, Gerald WL, Qin J, Cheung NK. Molecular genetics of neuroblastoma and the implications for clinical management: A review of the MSKCC experience. Oncologist 2001; 6(3)263–8
- Lastowska M, Cullinane C, Variend S, Cotterill S, Bown N, O'Neill S, et al. Comprehensive genetic and histopathologic study reveals three types of neuroblastoma tumors. J Clin Oncol 2001; 19(12)3080–90
- Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garrin E, Jallas AC, Combaret V, et al. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004; 6(6)625–30
- Rettig WJ, Spengler BA, Chesa PG, Old LJ, Biedler JL. Coordinate changes in neuronal phenotype and surface antigen expression in human neuroblastoma cell variants. Cancer Res 1987; 47(5)1383–9
- Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 2005; 433(7023)317–22
- Sarkar L, Sharpe PT. Expression of Wnt signalling pathway genes during tooth development. Mech Dev 1999; 85(1–2)197–200
- Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 2004; 131(22)5639–47
- Yang Y. Wnts and wing: Wnt signaling in vertebrate limb development and musculoskeletal morphogenesis. Birth Defects Res C Embryo Today 2003; 69(4)305–17
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398(6726)422–6
- Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74(5)1043–50
- Clapper ML, Coudry J, Chang WC. beta-catenin-mediated signaling: A molecular target for early chemopreventive intervention. Mutat Res 2004; 555(1-2)97–105
- Lin SY, Xia W, Wang JC, Kwong KY, Sphon B, Wen Y, et al. Beta-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A 2000; 97(8)4262–6
- Roberts LR, Gores GJ. Hepatocellular carcinoma: Molecular pathways and new therapeutic targets. Semin Liver Dis 2005; 25(2)212–25
- Kurihara T, Ikeda S, Ishizaki Y, Fujimori M, Tokumoto N, Hirata Y, et al. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid 2004; 14(12)1020–9
- Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. Beta-catenin dysregulation in thyroid neoplasms: Down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol 2001; 158(3)987–96
- Heathcott RW, Morison IM, Gubler MC, Corbett R, Reeve AE. A review of the phenotypic variation due to the Denys-Drash syndrome-associated germline WT1 mutation R362X. Hum Mutat 2002; 19(4)462
- Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982; 31(1)99–109
- Park WS, Oh RR, Park JY, Lee SH, Shin MS, Kim YS, et al. Frequent somatic mutations of the beta-catenin gene in intestinal-type gastric cancer. Cancer Res 1999; 59(17)4257–60
- Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene 1999; 18(47)6615–20
- Moreno-Bueno G, Gamallo C, Perez-Gallego L, Contreras F, Palacios J. beta-catenin expression in pilomatrixomas. Relationship with beta-catenin gene mutations and comparison with beta-catenin expression in normal hair follicles. Br J Dermatol 2001; 145(4)576–81
- Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet 2002; 31(3)306–10
- James LF. Teratological research at the USDA-ARS poisonous plant research laboratory. J Nat Toxins 1999; 8(1)63–80
- Keeler RF. Cyclopamine and related steroidal alkaloid teratogens: Their occurrence, structural relationship, and biologic effects. Lipids 1978; 13(10)708–15
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425(6960)846–51
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 2000; 14(8)994–1004
- Rubin JB, Rowitch DH. Medulloblastoma: A problem of developmental biology. Cancer Cell 2002; 2(1)7–8
- Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nat Rev Cancer 2002; 2(5)361–72
- Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003; 422(6929)313–7
- Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, Scharwachter C, et al. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol 2005; 152(1)43–51
- Taylor MD, Zhang X, Liu L, Hui CC, Mainprize TG, Scherer SW, et al. Failure of a medulloblastoma-derived mutant of SUFU to suppress WNT signaling. Oncogene 2004; 23(26)4577–83
- Grego-Bessa J, Diez J, Timmerman L, de la Pompa JL. Notch and epithelial-mesenchyme transition in development and tumor progression: Another turn of the screw. Cell Cycle 2004; 3(6)718–21
- Harper JA, Yuan JS, Tan JB, Visan I, Guidos CJ. Notch signaling in development and disease. Clin Genet 2003; 64(6)461–72
- Maillard I, Pear WS. Notch and cancer: Best to avoid the ups and downs. Cancer Cell 2003; 3(3)203–5
- Shawber C, Nofziger D, Hsieh JJ, Lindsell C, Bogler O, Hayward D, et al. Notch signaling inhibits muscle cell differentiation through a CBF1-independent pathway. Development 1996; 122(12)3765–73
- Lewis J. Notch signalling and the control of cell fate choices in vertebrates. Semin Cell Dev Biol 1998; 9(6)583–9
- Borowski C, Martin C, Gounari F, Haughn L, Aifantis I, Grassi F, et al. On the brink of becoming a T cell. Curr Opin Immunol 2002; 14(2)200–6
- Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306(5694)269–71
- Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet 2003; 33(2)208–13
- Radtke F, Raj K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor?. Nat Rev Cancer 2003; 3(10)756–67
- Axelson H. The Notch signaling cascade in neuroblastoma: Role of the basic helix-loop-helix proteins HASH-1 and HES-1. Cancer Lett 2004; 204(2)171–8
- Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 2004; 64(21)7787–93
- Sriuranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res 2001; 61(7)3200–5
- Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003; 33(3)416–21
- Weijzen S, Zlobin A, Braid M, Miele L, Kast WM. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J Cell Physiol 2003; 194(3)356–62
- Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 2004; 64(19)6854–7
- Houde C, Li Y, Song L, Barton K, Zhang Q, Godwin J, et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 2004; 104(12)3697–704
- Labbe JC, Maddox PS, Salmon ED, Goldstein B. PAR proteins regulate microtubule dynamics at the cell cortex in C. elegans. Curr Biol 2003; 13(9)707–14
- Riechmann V, Gutierrez GJ, Filardo P, Nebreda AR, Ephrussi A. PAR-1 regulates stability of the posterior determinant Oskar by phosphorylation. Nat Cell Biol 2002; 4(5)337–42
- Wei X, Cheng Y, Luo Y, Shi X, Nelson S, Hyde DR. The zebrafish Pard3 ortholog is required for separation of the eye fields and retinal lamination. Dev Biol 2004; 269(1)286–301
- Sierko E, Tokajuk P, Zimnoch L, Wojtukiewicz MZ. The location of components of fibrinolytic system in laryngeal cancer]. Pol Merkuriusz Lek 2003; 15(85)81–5
- Kaufmann R, Junker U, Nuske K, Westermann M, Henklein P, Scheele J, et al. PAR-1- and PAR-3-type thrombin receptor expression in primary cultures of human renal cell carcinoma cells. Int J Oncol 2002; 20(1)177–80
- Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR-1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005; 120(3)303–13
- Jin E, Fujiwara M, Pan X, Ghazizadeh M, Arai S, Ohaki Y, et al. Protease-activated receptor (PAR)-1 and PAR-2 participate in the cell growth of alveolar capillary endothelium in primary lung adenocarcinomas. Cancer 2003; 97(3)703–13
- Liu Y, Gilcrease MZ, Henderson Y, Yuan XH, Clayman GL, Chen Z. Expression of protease-activated receptor 1 in oral squamous cell carcinoma. Cancer Lett 2001; 169(2)173–80
- Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol 2004; 24(5)1990–9
- Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, et al. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 1998; 4(8)909–14
- Iversen LH, Okholm M, Thorlacius-Ussing O. Pre- and postoperative state of coagulation and fibrinolysis in plasma of patients with benign and malignant colorectal disease–a preliminary study. Thromb Haemost 1996; 76(4)523–8
- Darmoul D, Gratio V, Devaud H, Lehy T, Laburthe M. Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol 2003; 162(5)1503–13
- Tellez C, McCarty M, Ruiz M, Bar-Eli M. Loss of activator protein-2alpha results in overexpression of protease-activated receptor-1 and correlates with the malignant phenotype of human melanoma. J Biol Chem 2003; 278(47)46632–42
- Zhang X, Hunt JL, Landsittel DP, Muller S, Adler-Storthz K, Ferris RL, et al. Correlation of protease-activated receptor-1 with differentiation markers in squamous cell carcinoma of the head and neck and its implication in lymph node metastasis. Clin Cancer Res 2004; 10(24)8451–9
- Darmoul D, Gratio V, Devaud H, Laburthe M. Protease-activated receptor 2 in colon cancer: Trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem 2004; 279(20)20927–34
- Shibata K, Yada K, Matsumoto T, Sasaki A, Ohta M, Kitano S. Protease-activating-receptor-2 is frequently expressed in papillary adenocarcinoma of the gallbladder. Oncol Rep 2004; 12(5)1013–6
- Miyata S, Koshikawa N, Yasumitsu H, Miyazaki K. Trypsin stimulates integrin alpha(5)beta(1)-dependent adhesion to fibronectin and proliferation of human gastric carcinoma cells through activation of proteinase-activated receptor-2. J Biol Chem 2000; 275(7)4592–8
- Shimamoto R, Sawada T, Uchima Y, Inoue M, Kimura K, Yamashita Y, et al. A role for protease-activated receptor-2 in pancreatic cancer cell proliferation. Int J Oncol 2004; 24(6)1401–6
- Walsh DC, Kakkar AK. Thromboembolism in brain tumors. Curr Opin Pulm Med 2001; 7(5)326–31
- Yamahata H, Takeshima H, Kuratsu J, Sarker KP, Tanioka K, Wakimoru N, et al. The role of thrombin in the neo-vascularization of malignant gliomas: An intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int J Oncol 2002; 20(5)921–8
- Kaufmann R, Zieger M, Tausch S, Henklein P, Nowak G. Meizothrombin, an intermediate of prothrombin activation, stimulates human glioblastoma cells by interaction with PAR-1-type thrombin receptors. J Neurosci Res 2000; 59(5)643–8
- Okamoto T, Nishibori M, Sawada K, Iwagaki H, Nakaya N, Jikuhara A, et al. The effects of stimulating protease-activated receptor-1 and -2 in A172 human glioblastoma. J Neural Transm 2001; 108(2)125–40
- Tsopanoglou NE, Maragoudakis ME. Role of thrombin in angiogenesis and tumor progression. Semin Thromb Hemost 2004; 30(1)63–9
- Wilson S, Greer B, Hooper J, Zijlstra A, Walker B, Quigley J, et al. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem J 2005; 388(Pt 3)967–72
- Wilson SR, Gallagher S, Warpeha K, Hawthorne SJ. Amplification of MMP-2 and MMP-9 production by prostate cancer cell lines via activation of protease-activated receptors. Prostate 2004; 60(2)168–74
- Vetterkind S, Boosen M, Scheidtmann KH, Preuss U. Ectopic expression of PAR-4 leads to induction of apoptosis in CNS tumor cell lines. Int J Oncol 2005; 26(1)159–67
- Galvez-Gastelum FJ, Sandoval-Rodriguez AS, Armendariz-Borunda J. Transforming growth factor-beta as a therapeutic target]. Salud Publica Mex 2004; 46(4)341–50
- Shepherd TG, Nachtigal MW. Identification of a putative autocrine bone morphogenetic protein-signaling pathway in human ovarian surface epithelium and ovarian cancer cells. Endocrinology 2003; 144(8)3306–14
- Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 2002; 7(12)1191–204
- Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, et al. Novel regulators of bone formation: Molecular clones and activities. Science 1988; 242(4885)1528–34
- Oxburgh L, Chu GC, Michael SK, Robertson EJ. TGFbeta superfamily signals are required for morphogenesis of the kidney mesenchyme progenitor population. Development 2004; 131(18)4593–605
- Hyatt BA, Shangguan X, Shannon JM. FGF-10 induces SP-C and Bmp4 and regulates proximal-distal patterning in embryonic tracheal epithelium. Am J Physiol Lung Cell Mol Physiol 2004; 287(6)L1116–26
- Munoz O, Fend F, de Beaumont R, Husson H, Astier A, Freedman AS. TGFbeta-mediated activation of Smad1 in B-cell non-Hodgkin's lymphoma and effect on cell proliferation. Leukemia 2004; 18(12)2015–25
- Lee DK, Kim BC, Brady JN, Jeang KT, Kim SJ. Human T-cell lymphotropic virus type 1 tax inhibits transforming growth factor-beta signaling by blocking the association of Smad proteins with Smad-binding element. J Biol Chem 2002; 277(37)33766–75
- Ro TB, Holt RU, Brenne AT, Hjorth-Hansen H, Waage A, Hjertner O, et al. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene 2004; 23(17)3024–32
- Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, et al. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. Br J Cancer 2002; 87(6)630–4
- Hardwick JC, Van Den Brink GR, Bleuming SA, Ballester I, Van Den Brande JM, Keller JJ, et al. Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 2004; 126(1)111–21
- Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Peterson GM, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001; 28(2)184–7
- Keller ET, Zhang J, Cooper CR, Smith PC, McCauley LK, Pienta KJ, et al. Prostate carcinoma skeletal metastases: Cross-talk between tumor and bone. Cancer Metastasis Rev 2001; 20(3–4)333–49
- Dai J, Kitagawa Y, Zhang J, Yao Z, Mizokami A, Cheng S, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res 2004; 64(3)994–9
- Haudenschild DR, Palmer SM, Moseley TA, You Z, Reddi AH. Bone morphogenetic protein (BMP)-6 signaling and BMP antagonist noggin in prostate cancer. Cancer Res 2004; 64(22)8276–84
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer 2004; 4(3)177–83
- Furney SJ, Higgins DG, Ouzounis CA, Lopez-Bigas N. Structural and functional properties of genes involved in human cancer. BMC Genomics 2006; 7(1)3
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995; 270(5235)467–70
- Fodor SP, Rava RP, Huang XC, Pease AC, Holmes CP, Adams CL. Multiplexed biochemical assays with biological chips. Nature 1993; 364(6437)555–6
- Mockler TC, Chan S, Sundaresan A, Chen H, Jacobsen SE, Ecker JR. Applications of DNA tiling arrays for whole-genome analysis. Genomics 2005; 85(1)1–15
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100(1)57–70
- Arribas J. Matrix metalloproteases and tumor invasion. N Engl J Med 2005; 352(19)2020–1r
- Paranko J, Pelliniemi LJ, Foidart JM. Epithelio-mesenchymal interface and fibronectin in the differentiation of the rat mesonephric and paramesonephric ducts. Differentiation 1984; 27(3)196–204
- Griffith CM, Hay ED. Epithelial-mesenchymal transformation during palatal fusion: Carboxyfluorescein traces cells at light and electron microscopic levels. Development 1992; 116(4)1087–99
- Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 2004; 166(3)359–67
- Ishii M, Merrill AE, Chan YS, Gitelman I, Rice DP, Sucov HM, et al. Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development 2003; 130(24)6131–42
- Duband JL, Monier F, Delannet M, Newgreen D. Epithelium-mesenchyme transition during neural crest development. Acta Anat (Basel) 1995; 154(1)63–78
- Hay ED, Zuk A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am J Kidney Dis 1995; 26(4)678–90
- Sorokin L, Sonnenberg A, Aumailley M, Timpl R, Ekblom P. Recognition of the laminin E8 cell-binding site by an integrin possessing the alpha 6 subunit is essential for epithelial polarization in developing kidney tubules. J Cell Biol 1990; 111(3)1265–73
- Wilson L, Maden M. The mechanisms of dorsoventral patterning in the vertebrate neural tube. Dev Biol 2005; 282(1)1–13
- Dyer MA, Farrington SM, Mohn D, Munday JR, Baron MH. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development 2001; 128(10)1717–30
- Black GC, Mazerolle CJ, Wang Y, Campsall KD, Petrin D, Leonard BC, et al. Abnormalities of the vitreoretinal interface caused by dysregulated Hedgehog signaling during retinal development. Hum Mol Genet 2003; 12(24)3269–76