Abstract
To select the MHC-I-binding epitope-rich sequence of mice telomerase reverse transcriptase (mTERT) and study the antitumor immune response induced by truncated TERT through mRNA-transfected dendritic cells (DCs) immunization in mice. The MHC-I-binding epitopes of TERT were predicted using bioinformatics software. The selected sequence of TERT (Truncated mTERT, TERTt, mTERT cDNA 1776 bp-2942 bp encoding 584 aa-969 aa) was cloned from B16 mouse melanoma cells and inserted into pBluescriptIIKS(+) plasmid downstream of the T7 promoter. TERTt RNA was prepared through in vitro transcription. Bone marrow-derived DCs were electroporated with TERTt RNA and used to immunize syngeneic naïve mice. The quantity and cytotoxic activity of TERT-specific cytotoxic T lymphocytes (CTLs) in mice spleen were evaluated using IFN-γ enzyme-linked immunospot (ELISPOT) and Lactate dehydrogenase release assay. The immunoprophylactic effects against TERT positive tumor induced by TERTt RNA transfected DC in vivo were evaluated through an immunized-challenged mouse model. TERTt was cloned and in vitro transcribed into TERTt mRNA. As shown in FCM analysis, the efficiency of DC electroporation is 35.1% (29.7–41.2%). After electroporation, a subtle increase of costimulator and MHC-II molecules were expressed on the cell surface. Immunization of TERTt mRNA transfected DCs induced IFN-γ-secreting CTLs which manifested specific cytotoxic activity against TERT-positive target cells. In a cancer mouse model, vaccination of TERTt mRNA-transfected DCs suppressed the growth of TERT positive tumors (p=0.001) and prolong the survival time of tumor-bearing animals (p=0.029). TERTt evokes an antitumor immune response in vivo which is targeted to TERT. TERTt can be used as an antigeneic sequence to produce anti-TERT tumor vaccine.
Telomeres are nucleoprotein complexes which cap the termini of all eukaryotic chromosomes and play essential roles in the maintenance of chromosomal integrity and cell viability. Telomerase, a specialized reverse transcriptase, synthesizes G-rich simple repeat sequences of telomeres de novo and maintains telomere length after each cell division cycle Citation[1], Citation[2].Reports of high telomerase activity in immortalized and cancer cells has led to the hypothesis that telomerase reactivation represents a critical step in the neoplastic process Citation[3], Citation[4]. Expression of telomerase in human cancer is directly linked to tumor growth and development, whereas inhibition of telomerase in telomerase-positive human tumors leads to growth arrest Citation[5], Citation[6]. Telomerase reverse transcriptase (TERT) is the rate-limiting component of enzyme telomerase. It was reported that hTERT levels are nearly undetectable in normal human cells, but are present in human tumor cell lines at 100-fold higher levels. As TERT amplification plays an important role in malignant transformation Citation[7–10],TERT is seen as an attractive candidate target for tumor therapy.
Vonderheide et al. Citation[11], Citation[12] identified the TERT-derived HLA-A2-binding epitope I540 (ILAKFLHWL) which triggers specific CD8+ CTLs to kill a wide range of hTERT+ tumors. He suggested that hTERT may be seen as a universal tumor associated antigen. There are constitutive expressions of telomerase RNA template in APC and the stable transfection of TERT might activate telomerase in APCs. Nair and his colleagues Citation[13] transfected dendritic cells with whole long TERT RNA and found that immunization of this DC vaccine can stimulated cytotoxic T lymphocytes (CTL) which lysed TERT+ tumor cells and inhibited the growth of TERT+ tumors in animals. As we well know, it is difficult to preserve RNA and transfect DC in a large scale because of the universal presence of RNAase in environment. Can we find an antigenic epitope-rich sequence in TERT? This sequence not only can serve as multivalent antigen but also can be manipulated conveniently in clinical setting. In our present study, using MHC-I Peptide Binding Predictions Citation[14], Citation[15], we retrieved the sequence of mTERT and found that there is accumulation of antigenic epitopes in the C-terminal part of TERT. We wanted to determine if immunization with this sequence can lead to immunoprophylactic effects against TERT-positive tumors. In the present study, we cloned truncated TERT (TERTt) cDNA into a plasmid vector and then performed in vitro transcription to obtain TERTt RNA. We transfected bone marrow-derived DC with TERTt RNA and studied the immune effects of this TERT sequence.
Materials and methods
Epitope prediction
The mouse TERT motif was loaded to predict MHC-I (H-2d and H-2b) restricted CTL epitopes using http://bimas.dcrt.nih.gov/molbio/hla_bind/. H-2Dd, H-2Kd, H-2Db and H-2Kb were selected as the MHC-I molecule analysis options. Predicted epitopes were limited to nonamer length.
Mice and cell lines
Six to 8-week-old female BALB/c mice (H-2d) and C57BL/6 mice (H-2b) were purchased from Laboratory Animal Research Center (LARC) of the Fourth Military Medical University (FMMU, Xi'an) and housed under pathogen-free conditions (License number: SCXK2002-005). All experiments involving the use of mice were performed in accordance with protocols approved by LARC. CT26 is a carcinogen-induced undifferentiated colon adenocarcinoma cell line from BALB/c mice. B16 is a mouse melanoma cell line from C57BL/6. Cells were maintained in RPMI-1640 medium (Gibco, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (FBS; Sijiqin Biotech, HangZhou), 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37°C, 50 ml/l CO2 and passaged every 2 days.
CT26 cells were transfected with pcDNA3.1(+)-mTERTt or pcDNA3.1(+) –hMAGEn by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The transfected CT26 cells were selected with 800 µg/ml G418 (Gibco, Carlsbad, CA) in culture medium and subcloned by limiting dilution assay in 96-well plates. The expanded cell clones were examined by PCR and RT-PCR using mTERTt or hMAGEn primers. The selected cell clones CT26-TERTt and CT26-hMAGEn were confirmed by immunofluorescence assay with anti-TERT antibody (BA0564, Boster Biotech) and anti-hMAGEn mouse serum, respectively.
Clone of TERTt and in vitro transcription assay
RNA was isolated from B16 cells using TRIZOL Reagent (Invitrogen, Carlsbad, CA). Total tumor RNA (1 µg) was reverse transcribed in a final volume of 20 µl using ThermoScript RT-PCR System with Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA). First-strand cDNA synthesis was primed with 1 µl of 50 mµ oligo-dT primer and the reaction was incubated at 55°C for 60 min. A total of 2 µl RT product was used to perform PCR reaction with TERTt primer: sense 5′-GACGAATTCGACATGGAGAGAGTGCGGCTACGGGAG-3′, antisense 5′-CACGTCGACCTACGACAGGAGCTTGTTCCGCAT-3′ (underline indicates the endonuclease cutting sites and start /end codon). The RT-PCR results were analyzed by 1% agarose gel electrophoresis and cloned into pBluescriptIIKS(+) (Promega) between EcoRI and SalI to construct pBluescriptIIKS(+)-TERTt. The sequence of TERTt was confirmed by DNA sequencing (Bioasia Inc.). The SacI-NotI fragment containing the EGFP coding region was isolated from pEGFP1 (Clontech, CA) and subcloned into the SacI-NotI site of pBluescriptIIKS(+) downstream of the T7 promoter to construct pBluescriptIIKS(+)-EGFP. The EcoRI-XhoI fragment encoding whole length of human MAGE-n Citation[16] was isolated from pcDNA3.1(+)-hMAGEn and used to construct pBluescriptIIKS(+)-hMAGEn.
The reconstructed plasmids were linearized to serve as DNA templates for in vitro transcription (IVT). IVT was performed by using mMessage mMachine T7 Ultra kit (Ambion, USA) according to the manufacturer's instructions. IVT RNA was purified by DNase I digestion followed by LiCl precipitation and 70% ethanol wash. For each experiment, at least three different batches of mRNA were used. RNA quality was confirmed by agarose-formaldehyde gel electrophoresis. RNA concentration was assayed by spectrophotometric analysis at OD260. RNA was stored at −80°C in 1 µg/µl aliquots.
Electroporation of murine bone marrow-derived DCs with RNA
Mouse bone marrow-derived DCs were generated as described by Lutz et al. Citation[17] with minor modifications Citation[18]. The harvest DCs were enumerated and resuspended in PBS at 5×106 cells/ml. Prior to electroporation, DCs were washed twice with serum-free Opti-MEM and resuspended to a final concentration of 107 cells/ml in Opti-MEM (Gibco BRL). A total of 0.5 ml of the DC suspension was mixed with 10 µg of IVT RNA and electroporated in a 0.4 cm-gap sterile disposable cuvette using the Gene PulserIIElectroporation System (Bio-Rad, USA). Cells were transfected with a voltage pulse of 300 V, with a capacitance of 150 µF for 6 milliseconds Citation[19]. The BALB/c or C57BL/6 derived DCs were electroporated with no RNA, TERTt mRNA or hMAGEn mRNA respectively, so as to generate blank DC, TERTt-DC and hMAGEn-DC. DCs were also eletroporated with EGFP mRNA to evaluate the transfection efficiency. After electroporation, fresh complete medium was added to the cell suspension immediately and cells were further incubated at 37°C in a humidified atmosphere supplemented with 50 ml/l CO2 for 24 h. The expression of costimulator CD86 and MHC-I molecule were analyzed by FCM (FACScan, Becton Dickinson) to confirm the phenotypic changes of DCs before and after electroporation. EGFP-transfected cells were checked for EGFP expression 24 h after electroporation by using flow cytometry (FCM) analysis. For EGFP analysis in DC cultures, CD11c positive cells were gated and then evaluated for EGFP expression.
Immunization-challenge assay
Naïve syngeneic mice were immunized by i.v. DC vaccine (blank DC, TERTt-DC, or hMAGEn-DC) through the tail vein at day 0 and day 7 with 5×105 DCs in 100 µl PBS. Seven days after second immunization, splenocytes of immunized mice were harvested and depleted of RBCs with ammonium chloride Tris buffer and were used as the effectors to perform ELISPOT and lactate dehydrogenase release assays Citation[20]. For tumor challenge experiment, 1×104 B16 cells were inoculated into the back of C57BL/6 mice subcutaneously and every group contained seven mice. The tumor volume was calculated by using the standard formula: V = a×b2/2, where ‘a’ is the longest superficial diameter and ‘b’ is the shortest superficial diameter. Those were measured with vernier calipers at the 14th day after tumor cell inoculation. The mice were all fed to observe the tumor-bearing survival time.
Immunofluorescence assay
CT26 cell crawling cover slips were prepared in 6-well cell culture plate. The cells were fixed with cold methanol at −20oC for 5 min and permeabilized with 0.1% TritonX-100 in PBS for 5 min. Cells were blocked with 3% BSA in PBS for 30 min in a humidified chamber. After incubation in primary antibody (diluted 1:50) for 1 hour, the cells were incubated in 1:150 diluted FITC-conjugated secondary antibody for 1 hour. After mounting the coverslips with buffered glycerol, the cells were examined with fluorescence microscopy.
ELISPOT analysis
The murine interferon-gamma ELISPOT kit (Diaclone, France) was used to determine tumor-specific, IFN-γ secreting T cells Citation[21]. 96-well filtration plates (Nunc, Roskilde, Denmark) were coated with 100 µl capture antibody (clone DB1). After an overnight incubation at 4°C, the wells were washed and blocked with 2% dry skimmed milk in PBS. A total of 1×106 splenocytes isolated from the mice were added to the wells and incubated at 37°C, 50 ml/lCO2 for 20 h with 5×104 target cells (B16, CT26, CT26-TERTt and CT26-hMAGEn respectively) which had been pretreated with 50 mg/l mitomycin C in RPMI 1640 for 30 min. Plates were washed and incubated with 100 µl biotinylated polyclonal antibody at 37°C for 90 min. After the removal of unbound antibodies, 100 µl avidin-alkaline phosphatase was added and plates were incubated for 1 h at 37°C. After washing, spots were developed by adding 100 µl of ready-to-use BCIP/NBT buffer and incubated at room temperature for spot formation. The spots were scanned and counted.
Lactate dehydrogenase release assay
Specific cytolytic activities of murine splenocytes were determined by LDH release assay. The CT26, CT26-TERT, CT26-hMAGEn and B16 were used as target cells respectively. Effectors and target cells were mixed at serial E/T ratios of 5:1, 10:1, 20:1, 40:1 at 0.2 ml/well in 96-well round-bottomed plates (Nunc, Roskilde, Denmark). After incubation for 4 h, cells were centrifuged at 500 r/min and the cell-free supernatant was collected for LDH assay using the CytoTox96 kit (Promega, Madison, WI, USA). The percentage of specific LDH release was calculated by the following formula:% cytotoxicity=[(experimental LDH release) – (spontaneous LDH release by effector or target cells)/ (maximum LDH release)×(spontaneous LDH release)]×100. For the controls, the target cells were incubated either in culture medium alone to determine spontaneous release or in a mixture of 2% Triton X-100 to define the maximum LDH release. The spontaneous release was always <10% of the maximum release. All assays were performed in triplicate.
Statistical analysis
All experiments were repeated at least three times. Data were expressed as mean±SE. Tumor volume and immune cell yield data were analyzed by one-way analysis of variance (ANOVA). All analyses were conducted with SPSS8.0 software. Values of p < 0.05 were considered statistically significant.
Results
MHC-I-binding epitopes of TERT
The nonamer H-2Dd, H-2Kd, H-2Db and H-2Kb binding epitopes of C-terminal part of mouse telomerase reverse transcriptase were predicted as shown in .
Cloning and IVT of TERTt
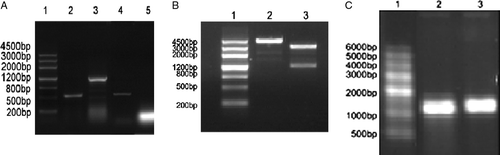
The expression of TERTt (mTERT cDNA 1776 bp-2942 bp encoding 584 aa-969 aa) can be detected in B16 cells but not in CT26 cells as shown in A. TERTt cDNA was cloned from B16 cells using RT-PCR and pBluescriptIIKS(+)-TERTt was constructed. Using linearized pBluescriptIIKS(+)-TERTt as template, in vitro transcription was performed to generate TERTt RNA (B&C). pBluescriptIIKS(+)-EGFP and pBluescriptIIKS(+)-hMAGEn were also constructed, linearized and the EGFP mRNA and MAGEn mRNA were all generated through in vitro transcription. DNA sequencing confirmed that the sequence of TERTt completely matches to the corresponding sequence of mouse TERT cDNA (NM 009354).
Figure 1. (A) The expression of TERTt in B16 and CT26 cells. 1: DNA marker I; 2: RT-PCR product of -actin in B16 cells; 3:RT-PCR product of TERTt in B16 cells; 4: RT-PCR product of -actin in CT26 cells; 5: RT-PCR result of CT26 cells using TERTt primers. (B) Linearization of pBluescriptIIKS(+)-TERTt : 1. DNA marker I 2. Linearized pBluescriptIIKS(+)-TERTt template 3.Double enzyme digestion of pBluescriptIIKS(+)-TERTt. (C) In vitro transcription of TERTt mRNA: 1. RNA marker 2.Capped TERTt mRNA 3. Capped TERTt mRNA with poly-A tail.
RNA electroporation of DC
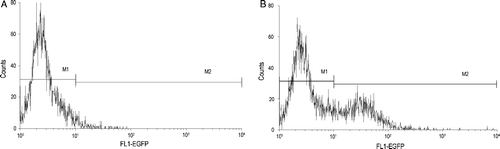
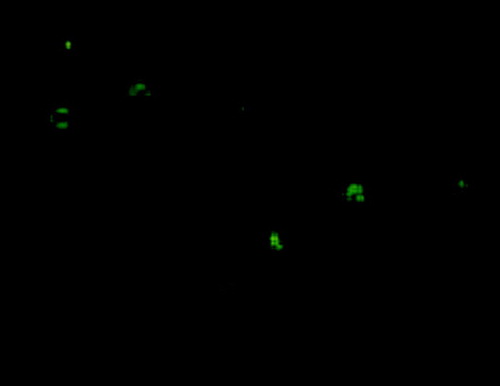
DC showed higher EGFP expression after EGFP RNA electroporation. FCM analysis directly shows that the EGFP RNA transfection efficiency of CD11c+ DC is as much as 35.1% (29.7–41.2%, five different eletroporation) at 24 h post electroporation (). DC surface molecules were measured by staining with PE conjugated-anti-mouse CD11c (N418, hamster IgG), FITC conjugated-anti-mouse I-A/I-E (m5/114.15.2, ratIgG2b), FITC conjugated-anti-mouse CD86 (B7.1, PO3, rat IgG2b) and analyzed by FACS. There was little increase of CD86 and I-A/I-E expression at 24 h after TERTt-RNA electroporation. After electroporated with TERTt mRNA 24 h, the TERTt expression in DCs can be detected using immunofluorescence assay as showed in .
Figure 2. DCs were eletroporated with EGFP mRNA to evaluate the transfection efficiency. Post EGFP mRNA electroporation 24h, EGFP expression can be detected in CD11c+ DCs using FCM analysis. (A) The DCs of 8th day culture system showed no EGFP expression; (B) After EGFP mRNA electroporated for 24h, The DCs of 7th day culture system showed EGFP expression.
TERTt-DC induced TERT-specific CTL response
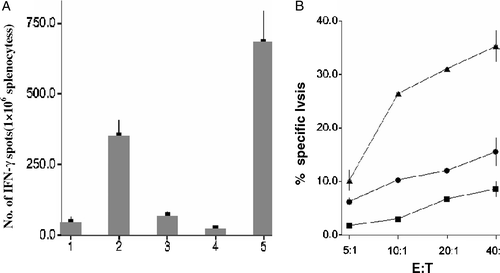
B16 is a natural TERT positive tumor cell line of C57BL/6 mouse. To investigate whether TERTt-DC have the potential to induce TERT-targeting antitumor effect, the splenocytes of different DC vaccine immunized mice were isolated to perform an IFN-γEliSpot experiment. B16 cells can stimulated a large number of IFN-γ-secreting T cells in TERTt-DC-immunized mice, but not in blank DC or hMAGEn-DC immunized mice. Compared to blank DC and hMAGEn -DC, TERTt-DC primed splenocytes displayed the strongest cytotoxic activity against B16 cells ().
Figure 3. TERTt mRNA transfected DCs immunization can induce B16 specific CTLs in C57B/L mice. (A) TERTt mRNA transfected DC immunization can induce IFN-γ secreting specific Splenocytes responding to Mitomycin C treated B16 cells. 1: Splenocytes of Blank DC immunized mice; 2: Splenocytes of TERTt-DC immunized mice; 3: Splenocytes of hMAGEn-DC immunized mice; 4: Negtive control (Splenocytes of Blank DC immunized mice, no target). 5: Positive control (Splenocytes of Blank DC immunized mice, no target, 1mg/L PHA stimulated). (B) Splenocytes obtained from TERTt mRNA transfected DCs immunized mice showed escalated cytotoxic activity against Mitomycin C treated B16 cells at E:T ratios indicated. ▴: Splenocytes of TERTt-DC immunized mice; •: Splenocytes of Mn-DC immunized mice; ▪: Splenocytes of Blank-DC immunized mice. (Values are the mean±SD of triplicate wells)
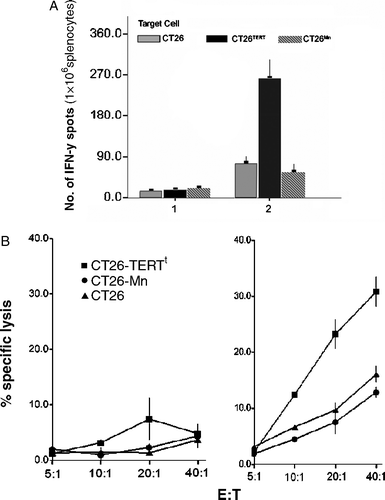
Primary CT26 cells showed undetectable TERT activity, we further transfected CT26 cells with pcDNA3.1(+)-TERTt to generate a CT26-TERTt cell line. After stimulated with target cells for 24 h, splenocytes derived from TERTt-DC-immunized mice only menifested IFN-γsecreting response against CT26-TERTt cells but not CT26 or CT26-hMAGEn cells. Furthermore, splenocytes of TERTt-DC-immunized mice displayed strong cytolytic activity against CT26-TERTt at an E:T ratio of 10:1, 20:1 and 40:1(A&B).
Figure 4. (A) Primary CT26 cells showed undetectable TERT activity. After stimulated with target cells for 24 hours, splenocytes derived from TERTt-DC immunized mice only menifested IFN-γ secreting response against CT26-TERTt cells but not CT26 or CT26-hMAGEn cells. 1: Splenocytes of Blank DC immunized mice; 2: Splenocytes of TERTt-DC immunized mice; (Values are the mean±SD of triplicate wells) (B) TERTt mRNA transfected DC immunization can generate TERTt specific CTLs in BALB/c mice in vivo. Splenocytes of TERTt-DC immunized mice displayed strong cytolytic activity against CT26-TERTt at E:T ratio of 10:1, 20:1 and 40:1. Left: Splenocytes of Blank DC immunized mice; Middle: Splenocytes of TERTt-DC immunized mice. (Values are the mean±SD of triplicate wells)
Vaccination of TERTt-DC suppress TERT positive tumor growth in vivo
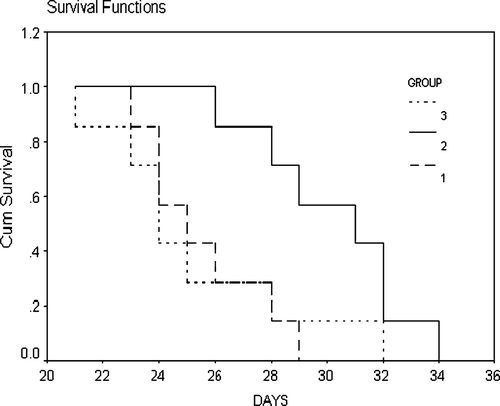
We examined whether TERTt-DC could induce TERT-targeting specific immunoprophylactic effects in vivo in an immunization-challenge mouse model. After being vaccinated with blank DC, TERTt-DC or hMAGEn-DC, respectively twice at an interval of 7 days, the mice received B16 cells inoculation subcutaneously. At day 14 post tumor inoculation, the tumor volume in blank DC vaccination group, TERTt-DC vaccination group and hMAGEn-DC vaccination group were 4456.4±378.6 mm3, 2121.4±381.1 mm3 and 3996.0±243.9 mm3 respectively. ANOVA analysis showed that there were significant differences in tumor volume among these three groups (p = 0.001). The B16 tumor volume in the TERTt-DC vaccination group is smaller than other two groups. The median tumor-bearing survival times of the blank DC, TERTt-DC, and hMAGEn-DC vaccination groups were 24 d, 31 d, and 25 d, respectively. Log-rank test showed that the median survival time was different in the three group of mice (p = 0.029). The mice vaccinated with TERTt-DC showed longer survival time than those vaccinated with blank DC or hMAGEn-DC ().
Figure 5. The Survival curve of different mRNA transfected DCs immunized/ B16-challenged mice. Log-rank test showed that the median survival time was different in these three groups of mice (p = 0.029). The mice vaccinated with TERTt-DC showed longer survival time than those vaccinated with Blank DC or hMAGEn-DC. 1: Blank DC immunized mice with B16 challenged; 2: TERTt-DC immunized mice with B16 challenged; 3: hMAGEn-DC immunized mice with B16 challenged.
Discussion
In recent years, TERT has been used as a universal tumor-associated antigen to evoke antitumor immune responses. Using a bioinformatic prediction scoring system, we predicted the 9-mer MHC-I binding epitopes of murine TERT and revealed that c-terminal partial sequence of mTERT holds many antigenic epitopes which can be presented through MHC-I pathway (H-2d or H-2b) by APCs. It's reported that RNA-loaded DC is a rapid and efficient measure to study the potential of tumor associated antigens to stimulate MHC-restricted anti-tumor immunity Citation[22–26]. In our present study, we cloned truncated TERT (TERTt) which encods 584aa-969 aa of mTERT and evaluated the antigenic nature of truncated TERT using TERTt mRNA-transfected DC.
Image1. The TERTt expression can be detected in Dendritic cells by using immunofluorescence assay 24 hours post TERTt mRNA electroporation. (First antibody: rabbit anti mouse TERT; Second antibody: FITC-goat anti rabbit IgG)
We cloned the TERTt cDNA and transfected DC with in vitro transcribed TERTt mRNA using electroporation. Using EGFP as an indicator, RNA electroporation efficiency of DC is estimated as 35.1%. We found a slight increase of MHC-I and CD86 expression of DCs post electroporation, which is similar to a result of Kalady's study. Since tumor-specific CTLs are the most important effectors in the antitumor response, we evaluated the specific recognition and cytolytic characteristics of mice splenic CTLs primed with TERTt RNA-transfected DC. After immunizing twice with TERTt RNA-transfected DC, the mice splenocytes showed targeting recognition and cytolytic activity to TERT-positive B16 cells. Furthermore, the splenocytes only reponded to and lyse TERTt transfected CT26 cells but not TERT negative CT26 cells. We also tested the in vivo immunoprophylactic effects of TERTt RNA-transfected DC using a vaccination-challenge model. We showed that pre-immunization with TERTt RNA-transfected DC inhibit the growth of B16 tumor and prolongs the tumor-bearing survival time of B16-challenged mice. Therefore, we conclude TERTt can evoke a TERT-targeting antitumor immune response in in vivo and TERTt might be used as an antigenic sequence to produce anti-TERT tumor vaccine.
It has been exhibited that mice express a higher basal level of telomerase activity in normal tissues compared to humans. Constitutive expression of TERT in mice might trigger a certain degree of immunotolerance to TERT vaccination. In humans, TERT expression is about 100-fold higher in tumor tissue than normal somatic tissue Citation[27]. The absence or lower concentration of basal TERT expression in human tissue may not trigger tolerance as much as in mouse. Thus, truncated TERT may have more potential in stimulating anti-TERT effects in humans. Further studies should be done to determine whether this truncated TERT vaccine could be pursued for humans.
It will be necessary to compare truncated TERT with the residual TERT and whole long TERT for evoking antitumor immune responses. TERTt can be conjugated with other immune enhancing sequences such as endosomal targeting motifs to improve presentation by APCs, or co-transfected with other antigenic sequences to extend the antitumor spectrum. It can also be loaded to APCs through reconstructed adenovirus transfection to facilitate the MHC-I pathway epitope presentation. Since TERT is activated in proliferating cells such as activated T and B cells, hematopoietic progenitors, germ cells, intestinal and liver cells Citation[28], Citation[29], pathological observations should be performed in TERTt-DC immunized mice to ensure that no autoimmune responses are present in these tissues and organs.
Acknowledgements
This research was supported by the Natural Science Foundation of Shaanxi Province, No. 2004C271
References
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985; 43: 405–13
- Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989; 59: 521–9
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266(5193)2011–5
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer 1997; 33: 787–91
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature 1999; 400(6743)464–8
- Herbert B, Pitts AE, Baker SI, Hamilton SE, Wright WE, Shay JW, et al. Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA 1999; 96: 14276–81
- Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene 1998; 16: 1723–30
- Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H, et al. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet 1998; 18: 65–8
- Counter CM, Meyerson M, Eaton EN, Ellisen LW, Caddle SD, Haber DA, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 1998; 16: 1217–22
- Ramakrishnan S, Eppenberger U, Mueller H, Shinkai Y, Narayanan R. Expression profile of the putative catalytic subunit of the telomerase gene. Cancer Res 1998; 58: 622–5
- Vonderheide RH, Hahn WC, Schultze JL, Nadler LM. The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity 1999; 10: 673–9
- Vonderheide RH, Domchek SM, Schultze JL, George DJ, Hoar KM, Chen DY, et al. Vaccination of cancer patients against telomerase induces functional antitumor CD8+ T lymphocytes. Clin Cancer Res 2004; 10: 828–39
- Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski JS, et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 2000; 6: 1011–7
- http://bimas.dcrt.nih.gov/molbio/hla_bind/.
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 1994; 152: 163–75
- Dong HL, Li ZS, Ye J, Qu P, Huang YY, Wu W, et al. Identification of HLA-A2-restricted CTL epitope encoded by the MAGE-n gene of human hepatocellular carcinoma. Cancer Biol Ther 2004; 3: 891–8
- Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 1999; 223: 77–92
- Qiu J, Li GW, Sui YF, Song HP, Si SY, Ge W. Heat-shocked tumor cell lysate-pulsed dendritic cells induce effective anti-tumor immune response in vivo. World J Gastroenterol 2006; 12: 473–8
- Van Meirvenne S, Straetman L, Heirman C, Dullaers M, De Greef C, Van Tendeloo V, et al. Efficient genetic modification of murine dendritic cells by electroporation with RNA. Cancer Gene Ther 2002; 9: 787–97
- Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 2000; 60: 1028–34
- Miyahira Y, Murata K, Rodriguez D, Rodriguez JR, Esteban M, Rodrigues MM, et al. Quantification of antigen specific CD8+ T cells using an ELISPOT assay. J Immunol Methods 1995; 181: 45–54
- Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 1999; 10: 281–7
- Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 1996; 184: 465–72
- Kalady MF, Onaitis MW, Padilla KM, Emani S, Tyler DS, Pruitt SK. Enhanced dendritic cell antigen presentation in RNA-based immunotherapy. J Surg Res 2002; 105: 17–24
- Ponsaerts P, Van Tendeloo VF, Berneman ZN. Cancer immunotherapy using RNA-loaded dendritic cells. Clin Exp Immunol 2003; 134: 378–84
- Kim KW, Kim SH, Jang JH, Lee EY, Park SW, Um JH, et al. Dendritic cells loaded with exogenous antigen by electroporation can enhance MHC class I-mediated antitumor immunity. Cancer Immunol Immunother 2004; 53: 315–22
- Greenberg RA, Allsopp RC, Chin L, Morin GB, DePinho RA. Expression of mouse telomerase reverse transcriptase during development, differentiation and proliferation. Oncogene 1998; 16: 1723–30
- Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nat Rev Immunol 2002; 2: 699–706
- Hiyama K, Hirai Y, Kyoizumi S, Akiyama M, Hiyama E, Piatyszek MA, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol 1995; 155: 3711–5