To the Editor
Gliofibromas are rare central nervous system (CNS) tumors with a mixture of glial and fibroblastic elements. Only 27 cases have been reported (12-supratentorial, 7-infratentorial, 8-spinal cord) Citation[1–5]. Complete excision was curative for most while some progressed despite adjuvant treatment. These tumors do not figure in the World Health Organization (WHO) classification of CNS tumors Citation[6]. We report our experience with three cases showing varied clinical presentation and behavior, thus attempting to extend the spectrum of existing knowledge about these tumors.
Case 1
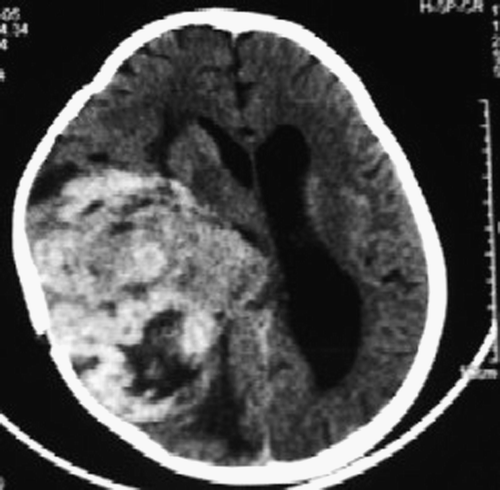
An 8-year old male presented with a 2-month history of headache, vomiting and complete blindness. He had earlier undergone gross total excision (GTE) for a right frontotemporal pilocytic astrocytoma and had been asymptomatic for 1 year. Examination revealed no light perception and papilledema. CT head showed a large temporoparietooccipital tumor with mass effect and midline shift (). Adjuvant radiotherapy (RT) (60 Gy over 6 weeks) and chemotherapy (6 cycles of Temozolomide (TMZ) 175 mg/m2 D1-5 q4 weeks) followed repeat tumor decompression. Subsequent evaluation till 1 year showed stable disease.
Figure 1. CT scan of head of patient 1 showing a large recurrent right temporoparietooccipital tumor with mass effect and midline shift.
Case 2
A 40-year old male presented with a history of generalized seizures, headache and vomiting for 3 months. MRI brain showed a right temporal lobe cystic mass (6×5 cm). Management comprised GTE and postoperative RT (56 Gy over 5½ weeks). He was disease-free at 3 years of follow-up.
Case 3
A 15-year old girl presented with persistent headache and projectile vomiting for 3 months. Imaging showed a 3×2 cm third ventricular tumor with obstructive hydrocephalus. She underwent a ventriculoperitoneal shunt insertion followed by GTE. Pathology revealed gliofibroma (grade II). She declined adjuvant treatment. Seventeen months later, a local recurrence (3.6×3.5 cm) was reexcised followed by adjuvant RT (56 Gy over 5½ weeks) and six cycles of TMZ 175 mg/m2 D1-D5 q4 weeks. She was disease-free at 2 years.
Resected tissue from all three cases was subjected to haematoxylin and eosin (H&E) and Gomori's reticulin staining, and immunohistochemistry using monoclonal antibodies to GFAP, vimentin, CD34, MIB-1 labeling index, cytokeratin (CK), epithelial membrane antigen (EMA), S-100, synaptophysin, neuron specific enolase (NSE), neurofilament protein and p53.
Except for the first specimen in case 1, microscopic examination was similar in all cases (primary disease in cases 2 and 3, recurrent disease in cases 1 and 3). Tumor was composed of two types of alternating areas, giving the marmoreal appearance. Glial areas were composed of sheets of cells, at places showing cord-like arrangement with moderate amounts of cytoplasm. No mitoses were seen. These cells were intensely positive for GFAP. The other areas were composed of spindle shaped reticulin-rich and GFAP-negative cells. In addition, case 3 also showed signet ring cells with adipocyte-like appearance (a–d,). Occasional inflammatory cell sprinkling was observed in the non-glial component. Both areas were negative for EMA, CK, CD34, synaptophysin and NSE. MIB-1 was <4% in all cases.
Figure 2. Photomicrograph of patient 3 showing islands of glial tissue alternating with fibroblastic stroma (a, H&E×100; b H&E×200). Another area showing fibroblastic tissue with adipose like areas (c, H&E×100) and at places showing whorling (d, H&E×100).
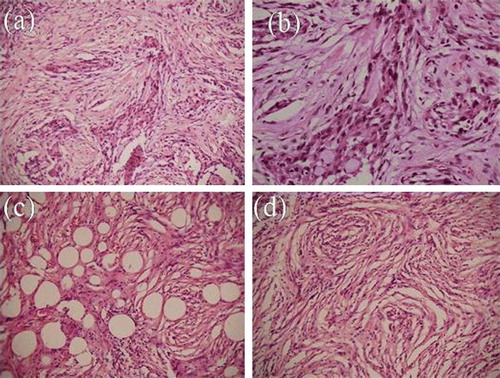
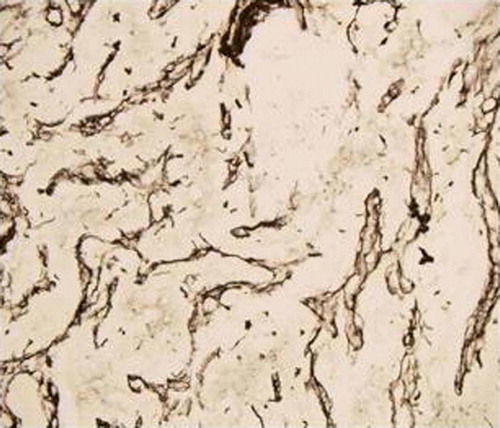
Figure 3. Photomicrograph of patient 3 showing reticulin positivity in stroma on immunohistochemistry (H&E×100)
Based on the above features, the possibility of gliofibroma, possibly WHO grade II, was considered.
Discussion
Tumors showing both glial and mesenchymal differentiation are uncommon in CNS. Gilosarcomas and sarcogliomas comprising malignant glial and sarcomatous elements are well recognized. In contrast, gliofibromas have a benign to malignant appearance on histopathology, depending on the degree of anaplasia of the glial element, the mesenchymal element being consistently benign. The cell of origin has been proposed to be endothelial, histiocytic, fibroblastic or a multipotent glial/mesenchymal progenitor Citation[7], Citation[8]. CT showing contrast enhancement, and isointense and hyperintense appearance on T1 and T2- weighted MRI respectively are characteristic. Differential diagnoses in adults include gliomas or gliosarcomas, and in children, desmoplastic astrocytomas and desmoplastic gangliogliomas Citation[3], Citation[9].
Friede first described a medullary gliofibroma in a 4-year old girl in 1978. Disease progression and death occurred within 8 months despite surgery, adjuvant RT and chemotherapy. Citation[1] Reported cases show presentation in the first two decades of life, no apparent gender predilection and mostly supratentorial tumors, though cerebellar and spinal cord tumors have also been seen. Our first two cases were supratentorial while the last had a third ventricular tumor.
Only primary tumors are described, except one which probably originated from hamartoma-like lesions Citation[10]. Our experience suggests that gliofbromas may originate either de novo (cases 2 and 3) or progress from benign tumors (case 1). The secondary lesion may either be similar to the primary tumor (case 3) or more aggressive (case 1). Degree of anaplasia of the glial component determines prognosis, as suggested by Sharma et al. who correlated MIB-1 and p53 in three cases to histology (benign vs. malignant) and clinical behavior (protracted vs. aggressive) Citation[11].
There are no clear-cut management guidelines. Benign tumors do well following GTE. Adjuvant chemotherapy and RT in partially resected or high grade lesions have been unsatisfactory. Suarez and coworkers reported an overall mortality rate of 23%, after adjuvant carboplatin and vincristine Citation[12]. All our patients received adjuvant RT. Response to adjuvant TMZ was encouraging in recurrent cases (cases 1 and 3).
We conclude that gliofibroma as a distinct entity warrants a place in the WHO brain tumor classification. Radiological features, extent of resection, MIB-1 and anaplasia may assist prognostication and decisions on adjuvant therapy. Additionally, we suggest that the use of TMZ may be extended to the adjuvant treatment of malignant or recurrent gliofibromas.
References
- Friede RL. Gliofibroma. A peculiar neoplasia of collagen forming glia-like cells. J Neuropathol Exp Neurol 1978; 37: 300–13
- Kim Y, Suh YL, Sung C, Hong SC. Gliofibroma: A case report and review of the literature. J Korean Med Sci 2003; 18: 625–9
- Erguvan-Onal R, Ates O, Onal C, Aydin NE, Kocak A. Gliofibroma: An incompletely characterized tumor. Tumori 2004; 90: 157–60
- Deb P, Sarkar C, Garg A, Singh VP, Kale SS, Sharma MC. Intracranial gliofibroma mimicking a meningioma: A case report and review of literature. Clin Neurol Neurosurg 2006; 108: 178–86
- Nomura M, Hasegawa M, Kita D, Yamashita J, Minato H, Nakazato Y. Cerebellar gliofibroma with numerous psammoma bodies. Clin Neurol Neurosurg 2006; 108: 421–5
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 2002; 61: 215–25
- Snipes GJ, Steinberg GK, Lane B, Horoupian DS. Gliofibroma. Case report. J Neurosurg 1991; 75: 642–6
- Schober R, Bayindir C, Canbolat A, Urich H, Wechsler W. Gliofibroma: Immunohistochemical analysis. Acta Neuropathol (Berl) 1992; 83: 207–10
- Serra A, Strain J, Ruyle S. Desmoplastic cerebral astrocytoma of infancy: Report and review of the imaging characteristics. AJR Am J Roentgenol 1996; 166: 1459–61
- Reinhardt V, Nahser HC. Gliofibroma originating from temporoparietal hamartoma-like lesions. Clin Neuropathol 1984; 3: 131–8
- Sharma MC, Gaikwad S, Mehta VS, Dhar J, Sarkar C. Gliofibroma: Mixed glial and mesenchymal tumour. Report of three cases. Clin Neurol Neurosurg 1998; 100: 153–9
- Suarez CR, Raj AB, Bertolone SJ, Coventry S. Carboplatinum and vincristine chemotherapy for central nervous system gliofibroma: Case report and review of the literature. J Pediatr Hematol Oncol 2004; 26: 756–60