Abstract
Introduction. In light to recently found contribution of factors associated with thrombosis and inflammation to carcinogenesis, we investigated the possible association of angiotensin I- converting enzyme (ACE) with increased risk for oral cancer. Materials and methods. In DNA samples of 160 patients with oral squamous cell carcinoma and 153 healthy controls of comparable ethnicity, age and sex, we studied the insertion/deletion (I/D) polymorphism in the ACE gene, which affects its transcription. Results. The I allele frequencies were significantly increased in patients compared to controls, 40.6% versus 27.5% (p < 0.001), respectively. The II homozygotes had a three-fold greater risk for developing oral cancer (odds ratio 3.17, 95% C.I. 1.32-7.61). A significant increase of I alleles was observed in patients regardless their smoking or alcohol consumption habits, early or advanced stage of cancer, presence or absence of a family history for cancer or thrombophilia (Fischer values p < 0.05). Discussion. These findings suggest that the I/D polymorphism, by affecting the ACE gene expression, is associated with the progress of oral oncogenesis.
Oral squamous cell carcinoma (OSCC) has a remarkable incidence worldwide and a fairly poor prognosis, which makes this disease a serious public health problem Citation[1]. Oral carcinogenesis is a multistep process occurring due to environmental factors such as smoking, alcohol abuse and betel quid chewing, gene alterations in oncogenes and tumor suppressor genes and viral infections Citation[2]. Recently, common polymorphisms in angiogenesis, inflammation and thrombosis-related genes have been associated with increased risk for oral cancer Citation[3–5]. One such factor related with both thrombosis and malignancies is angiotensin-converting enzyme (ACE) Citation[6–11].
ACE is the key enzyme in the rennin-angiotensin system (RAS), which is a circulatory cascade primarily involved in the regulation of blood pressure and serum electrolytes Citation[12]. ACE converts angiotensin I to angiotensin II, the major effector of the RAS Citation[12]. Angiotensin II is a potent vasoconstrictor and activator of aldosterone, induces reabsorption of sodium and raises the blood pressure Citation[12]. Therefore, ACE inhibitors are used in the treatment of heart failure, hypertension and coronary disease Citation[13]. Additionally ACE inactivates bradykinin, which is important in inhibiting cardiac hypertrophy, in limiting ischaemic myocardial injury and may also contribute to the biological behavior of tumors by stimulating growth and increasing vascular permeability Citation[14]. Actually, there is mounting evidence that ACE also participates locally in the pathology of several carcinomas, such as breast cancer, prostate cancer, endometrial cancer and oral precancerous lesions Citation[7–10]. It influences tumor cell proliferation, tumor cell migration, angiogenesis, and metastatic behavior Citation[12]. Inhibition of ACE activity suppresses tumor growth and angiogenesis in vitro and in vivo in animal models, while epidemiologic studies have provided evidence that ACE inhibitors may decrease the risk and mortality rate of cancer Citation[13], Citation[15].
The human ACE gene is located on chromosome 17q23 and expressed in a wide range of tissues including vascular endothelium, lung, kidney, heart and testes. It is also present in the circulation, although this may reflect cleavage of the enzyme by vascular endothelial surfaces Citation[12].
Segregation and linkage studies have provided strong support for an ACE-linked quantitative trait locus, although the underlying functional variants involved in predicting differences in ACE levels have not been fully defined Citation[16]. In European populations the majority of variation in ACE levels has been localized to an 18 kb region located between two ancestral recombination sites in the ACE gene Citation[16]. The high degree of linkage disequilibrium observed between several polymorphisms in this region has hampered efforts to disclose the functional variant for circulating ACE levels.
The best characterized factor which determines ACE activity is the insertion/deletion (I/D) polymorphism Citation[17]. This biallelic genetic polymorphism is characterized either by the absence (D allele) or the presence (I allele) of a 287 bp Alu repeat sequence inside intron 16 of the angiotensin I-converting enzyme gene, which results in the following three genotypes: II and DD homozygotes and ID heterozygotes. It seems to account for up to one-half of plasma ACE levels Citation[16]. Homozygotes for the I allele may display as low as half of the plasma ACE level compared to the homozygotes for the D allele, whereas the ID heterozygotes display an intermediate level Citation[16]. It has been assumed that the insertion fragment of DNA (I allele) may contain a silencer motif, since it has a similar sequence to other known suppressor genes (e.g. mouse rennin gene, human c-myc gene) or alternatively that I/D polymorphism has no functional activity and is in tight linkage disequilibrium with another variant around the ACE gene Citation[18]. The more common D allele frequency ranges in Europeans from 49 to 58%, while in Asians from 32 to 34%.
The presence of the D allele, by causing elevation in angiotensin II levels, could result in vasoconstriction and higher blood pressure. Evidence from animal models has suggested that angiotensin II stimulates neovascularization in normal tissue and neoplasms by promoting arteriolar smooth muscle cell proliferation Citation[19]. Captopril, a prototype ACE inhibitor, has been shown to inhibit proliferation in a variety of cell types, including cancer cells, and to reduce tumor growth in experimental models of cancer Citation[20]. ACE inhibitors are used widely as antihypertensive agents, and several epidemiological studies with conflicting results have examined cancer risks in long-term users of ACE inhibitors. One study suggested that users of ACE inhibitors exhibit a reduced risk of cancer when compared with general population, in contrast to other subsequent observational studies which failed to confirm the previously noted suggestion Citation[21], Citation[22].
Interestingly, several studies report association of ACE I/D polymorphism with several types of cancer such as breast cancer, prostate cancer, endometrial cancer and oral precancerous lesions Citation[7–10]. Intrigued by these observations, we aimed to investigate the putative significance of ACE levels in oral oncogenesis. In order to determine whether the ACE I/D polymorphism is associated with increased risk for oral cancer we investigated this polymorphism in patients with oral squamous cell carcinoma and healthy controls.
Materials and methods
The individuals under study were 313 Greeks and Germans, including 160 patients with oral squamous cell carcinoma and 153 healthy blood donors of equivalent age, ethnicity and sex. The patients were mostly men (n = 128, 80.0%) and their age ranged between 40–80 years (57.5±9.7 years) while the mean age for the whole group of patients was 58.5±10.1 years. The vast majority of the controls were males as well (n = 114, 74.5%) whose age varied between 38–83 years (56.2±10.6) while the mean age for the whole group of controls was 54.8±11.9 years.
The patients that were included in this study had already developed oral cancer and had been operated recently or up to a decade ago. In addition to clinical presentation, a biopsy with pathological diagnosis of tumor stages I-IV and a family history regarding any type of cancer or thrombosis were available for each patient. Fifty eight of them (36.3%) had one or two first degree relatives with cancer and their age range 41–83 years (58.7±10.4) did not differ significantly from the whole group of patients. Furthermore, 30 patients (18.8%) had one or two first-degree relatives with idiopathic thrombosis and an age range of 44–75 years (58.0±10.3), again with no statistical difference compared to the whole group. Fourteen patients (8.8%) had a positive family history for both cancer and thrombosis with a mean age of 56.1±8.6 years. Nearly all patients (95.0%) were smokers consuming at least ten cigarettes per day and about a third of them (32.5%) were ethanol abusers consuming three glasses of alcoholic drinks per day. Most of the studied individuals worked in a low-risk environment (with the exception of one patient and three controls who worked in chemical factories).
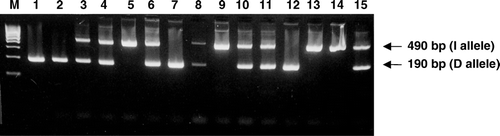
Blood samples were collected from all individuals after informed consent and DNA was isolated with the use of Nucleon™ kit (Amersham). Molecular detection of the ACE I/D polymorphism was performed by PCR amplification of intron 16. The presence (allele I) or absence (allele D) of the 287 bp Alu repeat sequence in the ACE gene was determined by gel electrophoretic evaluation of the size of PCR products. A 490 bp fragment indicated the presence of the I allele and a 190 bp fragment indicated the presence of the D allele (). The PCR conditions consisted of an initial denaturation step at 95°C for 5 min, followed by 31 cycles of 94°C for 55 s, 60°C for 1 min, and 72°C for 50 s, as well as a final elongation step at 72°C for 5 min. The primers used were sense: 5′-CTGGAGACCACTCCCATCCTTTCT-3′ and antisense: 5′-GATGTGGCCATCACATTCGTCAGAT-3′ Citation[10]. For verification of the results of molecular analysis some of the samples were tested twice, while additional molecular work was performed in order to confirm DD genotype samples by using a pair of primers (forward, 5′-TGGGACCACAGCGCCCGCCACTAC-3′; and reverse, 5′-TCGCCAGCCCTCCCATGCCCATAA-3′) that produce an amplified product only in the presence of the insertion Citation[10].
Figure 1. Determination of ACE I/D polymorphism genotypes by PCR amplification. The presence of a 490 bp fragment (I allele) and a 190 bp fragment (D allele) indicate the possible genotypes DD, ID and II. M: molecular weight marker; 1, 2, 7 and 12: DD homozygotes; 3, 4, 6, 8, 10, 11 and 15: ID heterozygotes; 5, 9, 13 and 14: II homozygotes.
The statistical analyses were performed using SAS® software (version 9.0; SAS Institute Inc.). The genotype distribution and allelic frequencies were analyzed by means of Fisher's exact test. The frequencies of genotypes of the whole group or subgroups of patients were compared to the respective genotypes of the control group. All statistical analyses concerning: number of relatives with a history of cancer, number of relatives with a history of thrombosis, nicotine abuse and alcohol abuse, have assumed that all controls have nil values for the above variables (i.e. all controls do not have a family history of cancer, all controls do not have a family history of thrombosis, all controls do not use tobacco, and all controls do not drink alcohol). Thus, odds ratios are most likely expected to overestimate the true likelihood of ACE genotypes and these variables. The age criterion for the adjustment of odds ratios has been set at 60 years. Similar frequency distributions regarding age are found in the respective genotypes between controls and patients. The Mantel-Haenszel method was used for the calculation of all odds ratios with a 95% confidence interval (CI). A p-value less than 0.05 was considered statistically significant.
Results
The prevalence of detected ACE I allele in healthy controls and patients with oral cancer are shown in . The data for the two tested populations (Greek and German healthy controls) were analyzed together, since there were no significant differences of allele frequencies of the I/D polymorphism among the two populations. For the ACE I/D polymorphism, genotype distributions and allele frequencies in the control group, in patients group and subgroups were as expected for a sample in Hardy-Weinberg equilibrium.
Table I. Prevalence of ACE I allele and carrier frequencies in healthy controls and patients with oral cancer. Statistical difference of frequencies observed in patients and their subgroups in comparison to controls (p-values). N.S.: not significant p-value.
Compared to the control group (genotypes II = 9, DD = 78, ID = 66), only the homozygotes for the I allele were significantly increased (p < 0.001) in the group of patients (genotypes II = 30, DD = 60, ID = 70). The same pattern of increased frequency of II homozygotes compared to the control group was also observed in the following patients’ subgroups a) patients with cancer stages I&II, genotypes II = 20, DD = 32, ID = 36 (p < 0.001), b) patients with cancer stages III&IV, genotypes II = 10, DD = 28, ID = 34, (p < 0.05), c) patients without family history of cancer, genotypes II = 25, DD = 40, ID = 36, (p < 0.001), d) patients without family history of thrombosis, genotypes II = 28, DD = 48, ID = 54, (p < 0.001), e) patients with tobacco abuse, genotypes II = 30, DD = 54, ID = 68, (p < 0.001), f) patients with alcohol abuse, genotypes II = 12, DD = 24, ID = 16, (p < 0.05) and g) patients without alcohol abuse, genotypes II = 18, DD = 36, id = 54 (p < 0.05). In the latter subgroup a significant increase in the ID heterozygotes was also observed, p < 0.05. Additionally, a significant increase only in the ID heterozygotes (p < 0.05) was found in the subgroup of patients with family history of cancer (genotypes II = 4, DD = 20, ID = 34). Finally, in the subgroups of patients with positive family history of thrombosis, along with the subgroup of patients without tobacco abuse no significant differences in either ID heterozygotes or II homozygotes were obtained (genotypes II = 2, DD = 12, ID = 16 and II = 0, DD = 6, ID = 2, respectively). In the latter subgroup, no safe conclusions can be drawn since the number of tested individuals was extremely small.
The observed I allele frequency in the control group was 27.5%, therefore similar to other European populations. The detected I allele and carrier frequencies, in the patients’ group and in every subgroup were significantly elevated in comparison to the control group (value p < 0.05), with the only exception being the non-smoking patients whose minimal number (n = 10) does not permit any safe conclusions (). Finally, there were no major differences in the frequencies of the two ACE alleles due to categorizations of sex, age, and age at onset of oral cancer.
Interestingly, compared to individuals with the DD genotype, the relative risk for oral squamous cell carcinoma for ID heterozygotes was 1.38 (95% CI 0.86–2.22), while for II homozygotes was even greater 4.33 (95% CI 1.94–9.66). Additionally, ID heterozygotes have a similar relative risk for developing early or advanced stages of oral cancer (1.33 and 1.44, respectively). For II homozygotes the respective relative risks are higher for stages I&II (5.42, 95% CI 2.26–12.95) than for stages III&IV (3.1, 95% CI 1.17–8.22).
Discussion
The angiotensin I-converting enzyme is a key factor in the production of angiotensin II and in the degradation of bradykinin, which are important peptides involved in cardiovascular physiology Citation[14]. This enzyme is a type I cell surface zinc metallopeptidase, expressed by many cell types of various organs and tissue types Citation[12]. It exerts its effects through the RAS system leading under normal conditions to vasoconstriction and therefore blood pressure increase Citation[12]. In addition, ACE influences tumor cell proliferation, tumor cell migration, angiogenesis, and metastatic behavior Citation[13].
An insertion/deletion (I/D) polymorphism in the ACE gene influences its transcription rate. Homozygotes II have about half of the plasma ACE level compared to the homozygotes DD, whereas the ID heterozygotes display an intermediate level Citation[16]. Several studies have reported association of the I/D polymorphism with increased risk for several types of cancer including oral precancerous lesions Citation[7–10].
In light of the above, in the present study the genotypes and allele frequencies of the ACE I/D polymorphism were investigated in a cohort of 160 patients with oral cancer in comparison to 153 healthy controls of equivalent age, sex and ethnicity. Despite the relatively small sample of studied individuals, the overall obtained data revealed an association of the I/D polymorphism with an increased risk for oral squamous cell carcinoma. Compared to the control group, the frequency of the low expression I allele was significantly increased in the whole group and practically every subgroup of patients.
Interestingly, the majority of studies that suggested an association of ACE I/D polymorphism with several types of cancer have reported increased prevalence of the high expression D allele in the cancer patients compared to the healthy controls Citation[7–10]. These studies have supported a tendency linking increased expression of ACE with cancer development by facilitating angiogenesis through higher production of angiotensin II. Angiotensin II induces vascular endothelial growth factor, which plays a pivotal role in tumor angiogenesis and correlates with aggressive behavior and poor prognosis Citation[23]. In addition, angiotensin II may enhance other cytokines and growth factors such as transforming growth factor β-1, platelet-derived growth factor and basic fibroblast growth factor that may be involved in neoplastic transformation Citation[23].
On the contrary, the results of this study suggest that in the oral region the ACE-related mechanism that leads to oral cancer development and progression may be quite different. A well established role of ACE, besides angiotensin II production, is the inactivation of bradykinin Citation[14]. Bradykinin is known to contribute to tumor formation through its ability to stimulate growth and increase vascular permeability Citation[14]. In light of the above, it may be suggested that the increased prevalence of the low expression I allele observed in our oral cancer patients would result in reduced inactivation of bradykinin and therefore, in this case oncogenesis could be driven through a bradykinin-related pathway and not through angiotensin II. This notion of a more sensitive bradykinin-related reaction in the oral cavity, in contrast to other tissues, is also supported by the fact that patients treated with ACE inhibitors often present with angioedemas exclusively in the oral region, due to the increased local levels in bradykinin Citation[24].
Interestingly, every study that revealed an association of the high expression D allele with cancer was performed in patients with sex hormone-related neoplasias Citation[7–9]. In these cancers, oestrogens increase hepatic synthesis of the renin substrate angiotensinogen, which is converted to angiotensin I, the substrate of ACE Citation[25]. This might indicate that in these particular cancers the increase in ACE due to the presence of the D allele acts in an accumulative manner to the already existing elevated ACE amounts due to oestrogens and a burst towards cancer formation occurs. Therefore, the ACE polymorphism may not be a predisposition factor by its own right for cancer development. In support to this notion, no association of the D allele was found with the development of sex hormone-unrelated cancers such as gastric carcinomas Citation[11].
To our knowledge, the only study investigating the possible association of the ACE I/D polymorphism with oral oncogenesis has been conducted on Taiwanese betel quid chewers presenting with oral precancerous lesions Citation[10]. Contrary to our findings, the Taiwanese study revealed an increase of the D allele in individuals with oral precancerous lesions in comparison to betel quid chewers with no such lesions Citation[10]. This conflicting observation may be due either to the relatively small sample of investigated individuals in the Asian study (n = 61 in each group) and/or the genetic difference of the two studied populations (Taiwanese against Europeans). Furthermore, oral precancerous lesions and oral squamous cell carcinoma are two different pathological entities, since only 4% of oral precancerous lesions lead to oral carcinoma; therefore their development could be controlled by different biological pathways.
In conclusion, the studied ACE polymorphism is associated with increased risk for oral cancer in certain individuals. As a consequence, it is of great importance to perform further genetic association studies regarding the contribution of additional factors related to angiogenesis, inflammation and thrombosis to oncogenesis in the oral region. Any positive findings could ultimately result in the undertaking of preventive measures safeguarding the health status and lives of certain at risk individuals in the general population.
Acknowledgements
This work was supported in part by EPEAEK “Pythagoras” grant 70/3/7391 of the Greek Secretariat of Research and Technology to E.V.
References
- Massano J, Regateiro F, Januario G, Ferreira A. Oral squamous cell carcinoma: Review of prognostic and predictive factors. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006; 102: 67–76
- Song C, Xing D, Tan W, Wei Q, Lin D. Methylenetetrahydrofolate reductase polymorphisms increase risk of esophageal squamous cell carcinoma in a Chinese population. Cancer Res 2001; 61: 3272–5
- Vairaktaris E, Yapijakis C, Kessler P, Vylliotis A, Ries J, Wiltfang J, et al. Methylenetetrahydrofolate reductase polymorphism and minor increase of risk for oral cancer. J Cancer Res Clin Oncol 2006; 132: 219–22
- Vairaktaris E, Yapijakis C, Serefoglou Z, Vylliotis A, Ries J, Nkenke E, et al. Plasminogen activator inhibitor-1 polymorphism is associated with increased risk for oral cancer. Oral Oncol 2006; 42: 888–92
- Vairaktaris E, Yapijakis C, Serefoglou Z, Derka S, Vassiliou S, Nkenke E, et al. The interleukin-8 (-251A/T) polymorphism is associated with increased risk for oral squamous cell carcinoma. EJSO (in press).
- Jackson A, Brown K, Langdown J, Luddington R, Baglin T. Effect of the angiotensin-converting enzyme gene deletion polymorphism on the risk of venous thromboembolism. Br J Haematol 2000; 111: 562–4
- Freitas-Silva M, Pereira D, Coelho C, Bicho M, Lopes C, Medeiros R. Angiotensin I-converting enzyme gene insertion/deletion polymorphism and endometrial human cancer in normotensive and hypertensive women. Cancer Genet Cytogen 2004; 155: 42–6
- Medeiros R, Vasconcelos A, Costa S, Pinto D, Lobo F, Morais A, et al. Linkage of angiotensin I-converting enzyme gene insertion/deletion polymorphism to the progression of human prostate cancer. J Pathol 2004; 202: 330–5
- Haiman C, Henderson S, Bretsky P, Kolonel L, Henderson B. Genetic variation in angiotensin I-converting enzyme (ACE) and breast cancer risk: The multiethnic cohort. Cancer Res 2003; 63: 6984–7
- Chung FM, Yang YH, Chen CH, Lin CC, Shieh TY. Angiotensin-converting enzyme gene insertion/deletion polymorphism is associated with risk of oral precancerous lesion in betel quid chewers. Br J Cancer 2005; 93: 602–6
- Rocken C, Lendeckel U, Dierkes J, Westphal S, Carl-McGrath S, Peters B, et al. The number of lymph node metastases in gastric cancer correlates with the angiotensin I-converting enzyme gene insertion/deletion polymorphism. Clin Cancer Res 2005; 11: 2526–30
- Stroth U, Unger T. The renin angiotensin system and its receptors. J Cardiovasc Pharmacol 1999; 33: 21–8
- Fujita M, Hayashi I, Yamashina S, Itoman M, Majima M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem Biophys Res Commun 2002; 294: 441–7
- Wu J, Akaike T, Hayashida K, Miyamoto Y, Nakagawa T, Miyakawa K, et al. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int J Cancer 2002; 98: 29–35
- Yoshiji H, Yoshii J, IkenakaY, Yoshiji H, Noguchi R, Yanase K, et al. Suppression of the renin-angiotensin system attenuates vascular endothelial growth factor-mediated tumor development and angiogenesis in murine hepatocellular carcinoma cells. Int J Oncol 2002;20:1227–31.
- McKenzie CA, Julier C, Forrester T, McFarlane-Anderson N, Keavney B, Lathrop GM, et al. Segregation and linkage analysis of serum angiotensin I-converting enzyme levels: Evidence for two quantitative-trait loci. Am J Hum Genet 1995; 57: 1426–35
- Zhu X, McKenzie CA, Forrester T, Nickerson DA, Broeckel U, Schunkert H, et al. Localization of a small genomic region associated with elevated ACE. Am J Hum Genet 2000; 67: 1144–53
- Yamada T, Horiuchi M, Morishita R, Zhang L, Pratt RE, Dzau VJ. In vivo identification of a negative regulatory element in the mouse rennin gene using direct gene transfer. J Clin Invest 1995; 96: 1230–7
- Fernandez LA, Twickler J, Mead A. Neovascularization produced by angiotensin II. J Lab Clin Med 1985; 105: 141–5
- Small W, Molteni A, Kim YT, Taylor JM, Chen Z, Ward WF. Captopril modulates hormone receptor concentration and inhibits proliferation of human mammary ductal carcinoma cells in culture. Breast Cancer Res Treat 1997; 44: 217–24
- Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, et al. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer?. Lancet 1998; 352: 179–84
- Friis S, Sorensen HT, Mellemkjaer L, McLaughlin JK, Nielsen GL, Blot WJ, et al. Angiotensin-converting enzyme inhibitors and the risk of cancer: A population-based cohort study in Denmark. Cancer 2001; 92: 2462–70
- Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Invest 2002; 82: 747–56
- Chiu AG, Krowiak EJ, Deeb ZE. Angioedema associated with angiotensin II receptor antagonists: Challenging our knowledge of angioedema and its etiology. Laryngoscope 2001; 111: 1729–31
- Harrap SB, Davidson HR, Watt G. The angiotensin I converting enzyme gene and predisposition to high blood pressure. Hypertension 1993; 21: 455–60