Abstract
The effects of ‘metronomic'or extended chemotherapy dosing schedules (ECS) are mediated through poorly understood anti-angiogenic mechanisms. ECS combined with biological anti-angiogenic agents have produced promising pre-clinical results. Materials and Methods. We have expanded the list of agents with an in vitro ECS profile to include the methylating agent temozolomide (Temodal®) and the anti-mitotic agent estramustine (Estracyt®). These agents were also combined with a specific anti-angiogenic inhibitor IMC-1C11 and a non-specific agent with anti-angiogenic properties, Compound 5h. The in vitro HUVEC ECS model system was optimised and cell proliferation assays undertaken. Results. As a single agent, estramustine inhibited endothelial cell proliferation with an IC50 of 4.5 µM and was active at 10–33% of the maximum tolerated dose (MTD) from clinical schedules, whilst temozolomide had IC50 of 6.6 µM and was active at 1–6% of MTD. In combination, significant synergy was seen with IMC-1C11 in combination with either drug, whilst modest additive effects were observed with Compound 5h. None of the combinations resulted in significant cytotoxicity or apoptosis. Discussion. The results show that ECS of temozolomide and estramustine can be significantly enhanced when combined with specific anti-angiogenic inhibitors in an in vitro HUVEC system.
‘Metronomic’ scheduling of chemotherapy is a clinical strategy whereby low-dose cycles of chemotherapeutic drugs are administered in a continuous or regularly-spaced fashion in order to inhibit tumour angiogenesis Citation[1]. Such a strategy represents a radical departure from the traditional norms of chemotherapy based on the maximum tolerated dose (MTD) paradigm, where the drug dose cycle is increased until dose-limiting toxicity is encountered. The potential benefits of ‘metronomic’ dosing over traditional schedules of chemotherapy include lower risk of acquiring drug resistance, lower incidence of systemic side effects and a lower degree of bone marrow perturbation – a process which can mobilize bone marrow-derived circulating endothelial progenitor cells which may contribute towards tumour angiogenesis Citation[2]. The ‘metronomic’ strategy is also suitable for combinations of cytotoxic drugs with new molecularly-targeted anti-angiogenic agents, and there is compelling pre-clinical evidence to suggest that anti-angiogenic efficacy is enhanced when cytotoxic drugs administered in ‘metronomic’ schedules are combined in this manner Citation[3], Citation[4]. Clinical trials are currently underway to confirm such pre-clinical findings Citation[5], Citation[6]. In addition, extended oral administration of chemotherapeutic agents appears to be the optimum schedule fulfilling all the major requirements of ‘metronomic’ dosing, and as such, cytotoxic drugs available through the oral route are likely to be the most suitable agents for this strategy.
However, many uncertainties remain concerning the principle of ‘metronomic’ dosing. Issues such as identifying the most suitable cytotoxic drugs, establishing the optimal dosing and frequency schedule, determining the biological activity of such therapy through surrogate markers, the potential development of acquired drug resistance in the long-term, all remain to be elucidated for each treatment strategy. At present there are some pre-clinical approaches that include animal and in vitro models of ‘metronomic’ dosing which are generally acceptable as useful for the study of this treatment strategy. The work we present in this study is based on one such approach Citation[7]. The previously published protocols of an in vitro endothelial model were used to investigate the potential anti-proliferative properties of estramustine and temozolomide both individually and in combination with specific and non-specific anti-angiogenic agents. The drugs represent different types of orally bioavailable agents which are in clinical use as conventional chemotherapeutic drugs at present. Estramustine is an anti-mitotic agent consisting of a conjugate of oestradiol and normustine and is at present licensed for the treatment of advanced hormone-refractory prostatic carcinoma. Temozolomide is an alkylating agent used in the treatment of melanoma and glioblastoma. Paclitaxel, an anti-mitotic drug which has proven efficacy in metronomic regimens either singly or in combination with anti-angiogenic agents in vitro and in vivo studies Citation[7], Citation[8] was used as a “positive control” cytotoxic agent and served as a comparison for estramustine and temozolomide.
We investigated the synergistic activity between a protracted dosing schedule and a specific anti-angiogenic agent (anti-VEGFR-2 monoclonal antibody, IMC-1C11) Citation[9] or a non-specific one (Compound 5h) Citation[10]. For the purposes of clarity we use the term metronomic only in a clinical context and prefer the term extended chemotherapy dosing-schedule (ECS) to describe the in vitro methodology. The role of anti-VEGFR-2 antibody in combinatorial ECS has been established in pre-clinical studies previously Citation[11], Citation[12]. Compound 5h is a novel orally bioavailable drug belonging to the FTI group of compounds for which there is growing evidence of anti-angiogenic properties Citation[13] through currently unknown mechanisms. The cell proliferation assays were conducted on dermal fibroblasts and human tumour cell lines for comparative purposes. In order to demonstrate the clinical relevance of all drugs in terms of possessing anti-proliferative properties at clinically achievable doses, the relevant ECS doses of each drug were correlated with its MTD using known data from conventional pharmacokinetic studies.
Materials and methods
Drugs
Purified temozolomide was kindly provided by Schering-Plough, UK; purified estramustine by Dr. B. Asp (Pharmacia, Sweden); Paclitaxel, (Sigma, UK); IMC-1C11 by Dr. D. Hicklin (ImClone, USA); and Compound 5h by Dr. I. Bell (Merck, USA). For in vitro experiments, all drugs were reconstituted at 10 mM by dissolving in DMSO. All drugs were diluted in culture medium immediately prior to use and the highest DMSO concentration in culture was less than 0.1% (v/v).
Cells and culture conditions
All media and tissue culture reagents used were purchased from Invitrogen (UK) unless otherwise stated. HUVECs were purchased from TCS Cellworks (UK). Human dermal fibroblasts and the human renal cell carcinoma cell line CAKI2 were purchased from the European Collection of Cell Cultures (UK); the human melanoma cell line A2058 was purchased from the American Type Culture Collection (LGC Promocell).
The HUVECs were maintained in Medium 199 with Earle's salts and Glutamax-I supplemented with 10% (v/v) heat-inactivated FBS (HiFBS), heparin (20 units/ml; Sigma, UK) and endothelial cell growth supplement (150 µg/ml; BD Biosciences, UK). Dermal fibroblasts were cultured in DMEM with non-essential amino acids, glucose and sodium pyruvate supplemented with glutamine (2 mM; Sigma, UK) and 10% (v/v) HiFBS. CAKI2 and A2058 cell lines were maintained in 10% (v/v) HiFBS McCoy's Modified 5A Medium with L-Glutamine and 10% (v/v) HiFBS RPMI 1640 Medium with L-Glutamine respectively. All cells were kept in a humidified atmosphere of 5% CO2 at 37°C and harvested with a solution of 0.25% (w/v) trypsin-0.03% (w/v) EDTA when they were in logarithmic phase of growth and maintained as described above. For the in vitro experiments, all cells used were below Passage 6.
Cell proliferation assay
In vitro chemosensitivity testing was performed on cells grown as a monolayer on 96-well flat-bottomed tissue culture plates (Sarstedt, USA). Each drug concentration was set up in quintuplicate. Preliminary experiments were conducted to determine the optimal cell seeding density which enabled the longest period of uninterrupted growth in 96-well plates up to confluence. HUVECs were seeded at 3 500 cells/well, while fibroblasts, CAKI2 and A2058 cell lines were seeded at 1 500 cells/well, 1 000 cells/well and 500 cells/well respectively, each in 100 µl of media. The longest growth period, which determined the duration of drug exposure for all cells, was 4 days. Preliminary experiments were then conducted to determine the therapeutic range of each drug. Cells were treated with estramustine (0–100 µM), temozolomide (0–100 µM), paclitaxel (0–2 nM), IMC-1C11 (0–50 µg/ml) or Compound 5h (0–100 µM). The chemotherapeutic drugs were then combined with either IMC-1C11 or Compound 5h at 25 µg/ml and 0.1 µM respectively. For all experiments, in order to maintain a constant concentration of the drugs, the media was replenished with fresh solutions containing the appropriate drug concentrations on a daily basis according to Bocci et al. Citation[7].
For the cell proliferation assay, a standard (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) (MTS) assay (Promega, UK) was performed Citation[14]. The assay was performed immediately prior to drug addition to determine the baseline number of cells prior to treatment, and at the end of the period of drug treatment. Briefly, 20 µl of MTS solution was added to each well containing 100 µl of media and the background-subtracted absorbance at 4 hours was recorded. Cell growth from baseline was expressed as percentage change in absorbance values. To calculate relative growth, the growth of control, untreated cells was taken as 100%, and the corresponding growth of treated populations of cells was expressed as a percentage of control growth Citation[14]. The concentration of drugs required to reduce cell growth by 50% (IC50) as compared with controls was calculated by nonlinear regression fit analysis of the mean values of the data points. All data represent the mean of three independent experiments.
Statistical analysis
The results of relative cell growth are expressed as mean values±standard error. The data was analysed using a two-sample t-test for populations of unequal variance to compare the effects of different drug doses and combinations. SPSS software version 11.0 (SPSS, Chicago, USA) was used for the statistical analysis. A confidence level of p≤ of 0.05 was considered statistically significant.
Quantification of drug interactions
Drug interactions in terms of anti-proliferative activity were quantified by the fractional inhibition method Citation[15] as follows:
If the fractional inhibition of cell growth or viability of each drug is expressed as i1 and i2 and the fractional inhibition achieved via a combination of the two drugs is expressed as i1+2, the following criteria applies: (1) additive inhibition occurs when i1+2=i1+i2; (2) synergistic inhibition occurs when i1+2>i1+i2; and (3) antagonism occurs when i1+2<i1+i2.
Results
Anti-proliferative activity of estramustine, temozolomide, paclitaxel and compound 5h at low doses
The range of cell numbers, which refers to the minimum and maximum cell numbers for each cell type within the experimental 4 day period, were as follows: 3 500–6 400 cells/well for HUVECs; 1 500–2 250 cells/well for fibroblasts; 1 000–2 200 cells/well for CAKI2 cells; and 500–2 150 cells/well for A2058 cells. The derived baseline control (i.e. untreated cells) growth rate of HUVECs, fibroblasts, CAKI2 and A2058 cells after the 4 days was therefore 80.4±3.6%, 49.9±2.6%, 120.5±7.8% and 330.3±16.4% respectively. The percent of growth of the untreated cells after the 4 days was normalized to 100%, and the growth of treated cells was expressed as a percentage of this base-line growth Citation[14].
For the treated cell population, all drugs except IMC-1C11 inhibited cell proliferation. HUVECs were significantly more sensitive to the anti-proliferative effects of all drugs in a dose-dependent manner. Estramustine (A) affected only HUVECs from 1 µM (p < 0.05) to 10 µM (p < 0.05), with a calculated IC50 of 4.5 µM. In contrast, fibroblasts were only inhibited with drug concentrations at >50 µM (p < 0.05), giving a calculated IC50 of 80.9 µM. Both CAKI2 and A2058 cell lines were only significantly inhibited at >100 µM (p < 0.05 in both cases) and an IC50 was not reached for either cell lines.
Figure 1. Effect of continuous and protracted (i.e. 4-day) exposure of Estramustine (A), Temozolomide (B), Paclitaxel (C), and Compound 5h (D) on in vitro cell growth to determine therapeutic drug ranges. The growth of untreated control cells was taken as 100% and the growth of treated populations of cells was expressed as % of this. Columns and bars represent mean values +/− SE, respectively. Negative growth figures indicate that cell number after treatment was lower than cell number prior to treatment. For relevant statistical analyses, refer to main text.
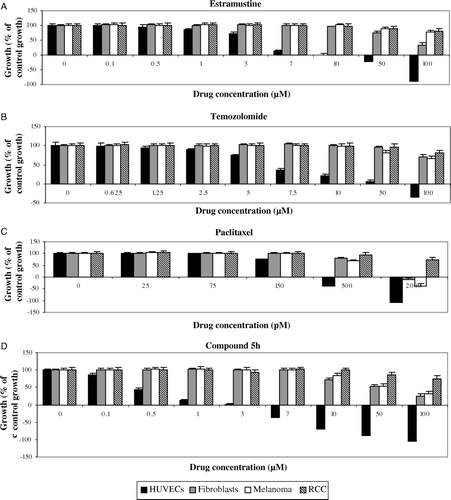
The same trend was observed with temozolomide, with HUVECs demonstrating significant sensitivity towards the drug at relatively low doses (B). Only HUVECs were inhibited from 2.5 µM (p < 0.05) to 10 µM (p < 0.05), with a calculated IC50 of 6.6 µM. In contrast, fibroblasts were only affected at >100 µM (p < 0.05), CAKI2 cell line >100 µM (p < 0.05) and A2058 cell line at >50 µM (p < 0.05). An IC50 was never achieved for these cells. For paclitaxel (C), only HUVECs were inhibited at 150 pM (p < 0.05), giving a calculated IC50 of 228 pM. Fibroblasts and A2058 cell line were only affected at >500 pM (p < 0.05 in both cases), giving IC50 values of 985 pM and 768 pM respectively. CAKI2 cell line was only significantly affected at 2 nM (p < 0.05) and an IC50 was not reached.
The findings that the ECS therapy with the conventional cytotoxic agents significantly more affected the low proliferative HUVECs compared to the highly proliferative CAKI2 and A2058 cells was not unexpected. As the investigated doses are very low they should not cause conventional cytotoxicity. The “metronomic” effects observed in the HUVECs may be mediated by the potent and endothelial specific angiogenesis inhibitor, thromdospondin-1 Citation[16] hence the tumourostatic effects are not due to the doses actually killing the cancer cells.
Compound 5h (D) showed endothelial-specific growth inhibitory activity, with only HUVECs significantly affected from 0.1 µM (p < 0.05) to 7 µM (p < 0.05), with a calculated IC50 of 0.44 µM. In contrast, fibroblasts and A2058 cell line were only inhibited at >10 µM (p < 0.05 in both cases), giving IC50 values of 54 µM and 58.7 µM respectively. CAKI2 cell line was only inhibited at >50 µM (p < 0.05) and an IC50 was not reached.
The monocloncal anti-VEGFR-2 antibody IMC-1C11 did not cause any significant inhibition of cell growth within the range of drug concentrations used (0–50 µg/ml).
Synergy of anti-proliferative activity of estramustine, temozolomide and paclitaxel at low doses in combination with IMC-1C11 or compound 5h
Although IMC-1C11 did not cause any significant inhibition of cell growth within the range of drug concentrations used (0–50 µg/ml), the effect of combination of the low dose chemotherapeutic agents with IMC-1C11 at a concentration of 25 µg/ml was investigated. The rationale for choosing this dose was provided by a previous study undertaken by Klement et al. Citation[12] who demonstrated that addition of IMC-1C11 (25 µg/ml) alone had no significant effect on HUVECs but when combined with the low dose cytotoxic agent, vinblastine, striking anti-mitotic effects were observed.
For estramustine, addition of 25 µg/ml IMC-1C11 (A) increased the growth inhibition of HUVECs, and there was synergistic inhibitory activity between 1–3 µM of estramustine (p < 0.05). Although synergy was not demonstrated with 25 µg/ml IMC-1C11 and 7 µM of estramustine, HUVEC growth was inhibited by 92.3% (A). The addition of 0.1 µM Compound 5h (A) to 7 µM of estramustine enhanced HUVEC growth inhibition, but this increase was lower than the expected combined inhibition of the two drugs based on single-drug schedules; hence neither synergy nor additive inhibitory activity was demonstrated between estramustine and Compound 5h. None of the other cell types were inhibited by single or combined drug schedules within the range of drug doses tested.
Figure 2. Effect of low-dose Estramustine (A), Temozolomide (B) and Paclitaxel (C) in combination with IMC-1C11 (25 µg/ml) on in vitro HUVEC growth compared with single-drug schedule, following continuous, 4-day exposure. The growth of untreated control cells was taken as 100% and the growth of treated populations of cells was expressed as % of this. Columns and bars represent mean values +/− SE, respectively. *p < 0.05 comparing combinational regime to single-drug schedule (synergistic effect).
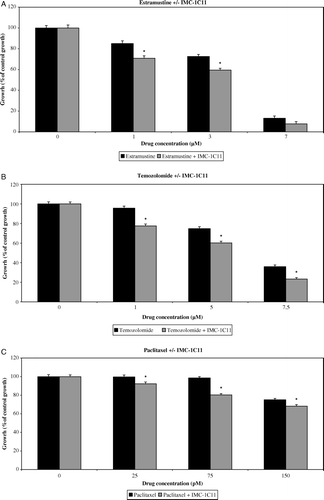
Figure 3. Effect of low-dose Estramustine (A), Temozolomide (B) and Paclitaxel (C) in combination with Compound 5h (0.1 µM) on in vitro HUVEC growth compared with single-drug schedule, following continuous, 4-day exposure. The growth of untreated control cells was taken as 100% and the growth of treated populations of cells was expressed as % of this. Columns and bars represent mean values +/− SE, respectively. *p < 0.05 comparing combinational regime to single-drug schedule (synergistic effect), †p < 0.05 comparing combinational regime to single-drug schedule (additive effect), ‡p < 0.05 comparing combinational regime to single-drug schedule.
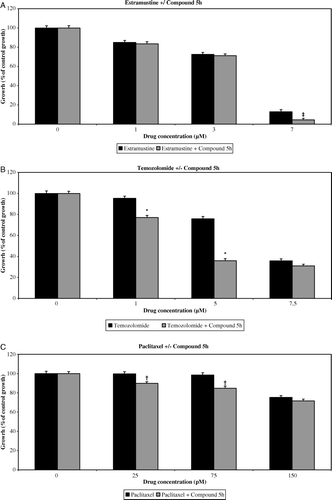
For temozolomide, addition of 25 µg/ml IMC-1C11 (B) increased HUVEC growth inhibition significantly, and synergistic inhibitory activity was demonstrated across all doses (p < 0.05 for all cases). A similar trend was observed with the addition of 0.1 µM Compound 5h to 1 µM and 5 µM of temozolomide (B), where significant synergy of growth inhibitory activity was demonstrated. However, there was no significant synergistic or additional inhibition at 7.5 µM of temozolomide with the addition of Compound 5h. Growth inhibition was not exhibited in the other cell types, either on single or combined drug schedules.
For paclitaxel, the addition of 25 µg/ml IMC-1C11 (C) significantly increased the growth inhibition of HUVECs (p < 0.05 for all cases), with synergy being demonstrated across all doses. Addition of 0.1 µM Compound 5h (C) resulted in additive inhibition rather than synergistic inhibition at 25 pM and 75 pM of paclitaxel respectively. Neither single-drug nor combined drug schedules incurred any growth inhibition in any of the other cell types within the range of drug doses tested.
In summary, modest but statistically significant synergy of endothelial-specific growth inhibitory activity was demonstrated between all cytotoxic agents and IMC-1C11 at low cytotoxic drug doses. Synergy was also demonstrated between temozolomide and Compound 5h, whereas paclitaxel and Compound 5h showed additive inhibitory activity rather than true synergy.
Discussion
We have shown that two orally bioavailable drugs belonging to different classes of cytotoxic compounds possess anti-endothelial properties at ECS dosing. Estramustine (an anti-mitotic agent) demonstrated an IC50 of 4.5 µM for HUVEC, but significant anti-proliferative effects were present at 1 µM. Pharmacokinetic data from Phase I clinical trials of oral and intravenous dosing schedules of estramustine suggest that levels of 1–3 µM are much lower than the peak plasma concentrations attained from maximum tolerated doses Citation[17], Citation[18]. In a Phase I trial of weekly-administered intravenous estramustine phosphate (EMP) carried out by Hudes et al. Citation[18], the MTD was determined to be 2 500 mg/m2. At a clinically well-tolerated dose of 2 000 mg/m2, the peak plasma concentrations (Cmax) of the active metabolites estramustine and estromustine exceeded 10 µM each. Even at a low dose of 500 mg/m2, the corresponding Cmax value of estramustine exceeded 2 µM. The results from the study also suggested that weekly intravenous EMP at 2 000 mg/m2 generated similar plasma concentrations of estramustine and estromustine as daily oral EMP at between 560–840 mg per day, which are the oral doses typically recommended for this drug. As such, these data and the findings from the present study suggest that estramustine may be suitable for use in a ‘metronomic’ schedule at 10–33% of its MTD.
Temozolomide (an alkylating agent) demonstrated an IC50 of 6.6 µM for HUVECs, but significant anti-proliferative effects were present at 2.5 µM. In Phase I clinical trials of temozolomide, daily oral 5-day schedules of the drug demonstrated a maximum tolerated dose of 250 mg/m2/day Citation[19]. At 50 mg/m2/day and 225 mg/m2/day, after 5 days of treatment, the Cmax values attained were 16 µM and 63.9 µM respectively. Consequently, there is some preliminary evidence from the present study to support the use of temozolomide in a ‘metronomic’ protocol at 1–5% of the MTD. In regard to the use of temozolomide in an extended continuous schedule, Brock et al. Citation[20] conducted a Phase I clinical trial which compared a 7-week daily oral schedule of temozolomide at a dose of 75 mg/m2/day for 7 weeks with a traditional 5-day regimen which comprised of a dose of 200 mg/m2/day repeated every 28 days in the treatment of patients with advanced melanoma or glioma. Although overall response rates in terms of tumour regression were similar, the extended dosing schedule resulted in a lower incidence of toxicities and a 2.1-fold greater drug exposure over a 4-week period. The anti-angiogenic effects of therapy were not assessed.
Paclitaxel demonstrated an IC50 of 228 pM for HUVECs, which was considerably higher than the corresponding value obtained by Bocci et al. Citation[7] which was 96 pM. Nevertheless, the same therapeutic specificity towards endothelial cells was demonstrated up to 1 nM. Previous pharmacokinetic studies involving paclitaxel have shown that 6-hour infusions and 24-hour infusions, each administered once every 3 weeks, resulted in an MTD of 250 mg/m2 and corresponding Cmax values of 13 µM and 1 µM respectively Citation[21]. Consequently, the data suggest that paclitaxel may be used at less than 1% of the MTD as part of a combined ‘metronomic’ protocol.
Preliminary studies using LDH release and annexin-V:FITC binding demonstrated no significant cytotoxicity or induction of apoptosis when studying the optimal drug concentrations (data not shown; available on request) although further investigations are required to clarify the exact mechanisms of action. Bocci et al. Citation[7] found that ECS scheduling with several cytotoxic agents including paclitaxel caused significant apoptosis in endothelial cells after 6 days of treatment. However, Browder et al. Citation[22] showed that low drug doses inhibited growth without inducing apoptosis when combining cyclophosphamide and TNP-470.
We have also presented evidence that suggests that the anti-endothelial effects of ECS chemotherapy can be further enhanced by the addition of molecular inhibitors of angiogenesis, such as anti-VEGFR-2 monoclonal antibodies or drugs which can potentially interfere with VEGFR-2 signalling pathways, such as FTIs (e.g. Compound 5h). Expression of the target for the anti-VEGFR-2 antibody (i.e. VEGFR-2) was confirmed on the HUVECs by flow cytometry (data not shown; available upon request) and our findings are in agreement with others. Citation[23], Citation[24]. Synergy of activity was demonstrated between all cytotoxic drugs studied and the anti-VEGFR-2 antibody. This finding correlates with the findings of Klement et al. Citation[10] involving the use of low-dose vinblastine and monoclonal anti-VEGFR-2 antibody to treat xenografts of neuroblastoma cell lines in SCID mice. Both vinblastine and anti-VEGFR-2 antibodies alone caused significant but transient tumour regressions, but the combination therapy resulted in full and sustained regressions lasting more than 6 months. The rationale for the combination was that since inhibition of VEGFR-2 can potentially prevent cells under cytotoxic stress from utilizing survival pathways to elude apoptosis, addition of anti-VEGFR-2 antibody to schedules comprising of low-doses of cytotoxic agents can be synergistic.
We also found that Compound 5h, a novel, orally bioavailable 3-aminopyrrolidinone FTI, demonstrated endothelial-specific anti-proliferative properties at very low doses and it appeared to have a wide endothelial-specific therapeutic window. FTIs were initially synthesized to inhibit the function of the ras protein, but their exact mechanism of action has since been the subject of much controversy, and may involve non-ras related processes Citation[25]. It is thought that one of the downstream pathways activated by the VEGF/VEGFR-2 system may involve the ras-dependent MAPK signalling pathway Citation[26], and this provides ras-inhibiting drugs such as FTIs with an anti-angiogenic rationale. Our study provides some evidence that FTIs may possess anti-endothelial properties, as previously shown by others Citation[27], Citation[28]. However, the effect of this non-specific anti-endothelial agent in combination with ECS chemotherapy is not as successful as that of combining ECS with a specific anti-angiogenic inhibitor in our in vitro model.
It has to be noted that the use of in vitro models based on HUVECs or any other endothelial cell type to represent tumour endothelium is not without its problems. There is a valid argument in using models based on cells of microvascular origin (e.g. human microvascular endothelial cells) to reflect more accurately the microvascular nature of angiogenesis in vivo. Furthermore it is patently obvious that the simplified demonstration of inhibitory effects on an endothelial cell model in vitro fails to take into account the anatomical complexity of even suboptimal tumour-related vasculature, which includes pericytes and other potential targets for anti-angiogenic effects. No in vitro model is truly representative of the in vivo process, and they should be regarded as approximations rather than true representations. Nevertheless, in vitro chemosensitivity assays do play crucial roles in the development of novel therapeutic strategies, especially within clinical trial settings Citation[29]. Our work demonstrates that in two classes of chemotherapy agents, the non-specific effects of ECS chemotherapy scheduling can be significantly enhanced when combined with an agent that targets the angiogenic pathway specific for the studied system. The combination however with a non-specific inhibitor of angiogenesis, even if this agent is effective as a single agent in the system studied, has unpredictable results. This may be related to the model used or the type of chemotherapy agents combined. In conclusion, further research needs to be undertaken in order to define the molecular mechanisms of action of the different anti-angiogenic agents to determine true synergistic responses.
In summary, based on the ECS protocol proposed by Bocci et al. Citation[7], we have demonstrated a readily available, easily accessible, rapid and technically simple in vitro system of screening potential cytotoxic drugs either alone or in combination for their anti-endothelial effects within an ECS schedule. Although in vitro assays of the type we have used cannot determine the mechanism of action, they can however afford some insight into what may be a clinically relevant combination of anti-angiogenics. In particular, they raise the issue that unless we improve our understanding of the mechanism of actions of the non-specific agents, we are bound to perpetuate the same problems of empiricism that have marked our use of MTD chemotherapy and its combinations.
Acknowledgements
We thank Schering-Plough (UK) for providing purified temozolomide; Dr. B. Asp (Pharmacia, Sweden) for providing purified estramustine; Prof. R. Kerbel and Dr. D. Hicklin (ImClone, USA) for providing IMC-1C11; and Dr. I. Bell (Merck, USA) for providing Compound 5h.
References
- Hanahan D, Bergers G, Bergsland E. Less is more, regularly: ‘Metronomic’ dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest 2000; 105: 1045–7
- Kerbel RS, Kamen BA. The anti-angiogenic basis of ‘metronomic’ chemotherapy. Nature Rev Cancer 2004; 4: 423–6
- du Manoir JM, Francia G, Man S, Mossoba M, Medin JA, Viloria-Petit A, et al. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res 2006; 12: 904–16
- Yap R, Veliceasa D, Emmenegger U, Kerbel RS, McKay LM, Henkin J, et al. Metronomic low-dose chemotherapy boosts CD95-dependent antiangiogenic effect of the thrombospondin peptide ABT-510: A complementation antiangiogenic strategy. Clin Cancer Res 2005; 11: 6678–85
- Buckstein R, Kerbel RS, Shaked Y, Nayar R, Foden C, Turner R, et al. High-dose celecoxib and metronomic “low-dose” cyclophosphamide is an effective and safe therapy in patients with relapsed and refractory aggressive histology non-Hodgkin's lymphoma. Clin Cancer Res 2006; 12: 5190–8
- Orlando L, Cardillo A, Ghisini R, Rocca A, Balduzzi A, Torrisi R, et al. Trastuzumab in combination with metronomic cyclophosphamide and methotrexate in patients with HER-2 positive metastatic breast cancer. BMC Cancer 2006; 15: 225–35
- Bocci G, Nicolaou KC, Kerbel RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 2002; 62: 6938–43
- Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anticancer Drugs 2003; 14: 13–9
- Posey JA, Ng TC, Yang B, Khazaeli MB, Carpenter MD, Fox F, et al. A phase I study of anti-kinase insert domain-containing receptor antibody, IMC-1C11, in patients with liver metastases from colorectal carcinoma. Clin Cancer Res 2003; 9: 1323–32
- Bell IM, Gallicchio SN, Abrams M, Beshore DC, Buser CA, Culberson JC, et al. Design and biological activity of (S)-4-(5-([1-(3-chlorobenzyl)–2–oxopyrrolidin-3-ylamino]methyl)imidazol-1-ylmethyl)benzonitrile, a 3-aminopyrroli- dinone farnesyltransferase inhibitor with excellent cell potency. J Med Chem 2001; 44: 2933–49
- Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 2000; 105: R15–R24
- Klement G, Huang P, Mayer B, Green SK, Man S, Bohlen P, et al. Differences in therapeutic indexes of combination ‘metronomic’ chemotherapy and an anti-VEGFR-2 antibody in multidrug-resistant human breast cancer xenografts. Clin Cancer Res 2002; 8: 221–32
- Han JY, Oh SH, Morgillo F, Myers JN, Kim E, Hong WK, et al. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J Natl Cancer Inst 2005; 97: 1272–86
- Johns TG, Luwor RB, Murone C, Walker F, Weinstock J, Vitali AA, et al. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc Natl Acad Sci USA 2003; 100: 15871–6
- Webb JL. Enzyme and metabolic inhibitors. Academic Press, New York 1963; 150–153
- Bocci G, Francia G, Man S, Lawler J, Kerbel RS. Thrombospondin-1, a mediator of the anti-angiogenic effects of low dose metronomic chemotherapy. Proc Natl Acad Sci USA 2003; 100: 12917–22
- Gunnarsson PO, Andersson SB, Johansson SA, Nilsson T, Plym-Forshell G. Pharmacokinetics of estramustine phosphate (Estracyt) in prostatic cancer patients. Eur J Clin Pharmacol 1984; 26: 113–9
- Hudes G, Haas N, Yeslow G, Gillon T, Gunnarsson PO, Ellman M, et al. Phase I clinical and pharmacologic trial of intravenous estramustine phosphate. J Clin Oncol 2002; 20: 1115–27
- Dhodapkar M, Rubin J, Reid JM, Burch PA, Pitot HC, Buckner JC, et al. Phase I trial of temozolomide (NSC 362856) in patients with advanced cancer. Clin Cancer Res 1997; 3: 1093–100
- Brock CS, Newlands ES, Wedge SR, Bower M, Evans H, Colquhoun I, et al. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res 1998; 58: 4363–7
- Rowinsky EK, Onetto N, Canetta RM, Arbuck SG. Taxol: The first of the taxanes, an important new class of antitumor agents. Semin Oncol 1992; 19: 646–62
- Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O'Reilly MS, et al. Anti-angiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 2000; 60: 1878–86
- Funovics P, Brostjan C, Nigisch A, Fila A, Grochot A, Mleczko K, et al. Effects of 15d-PGJ(2) on VEGF-induced angiogenic activities and expression of VEGF receptors in endothelial cells. Prostaglandins Other Lipid Mediat 2006; 79: 230–44
- Nimi Y, Mochida S, Matsui A, Inao M, Fujiwara K. PKC- and MAPK-independent upregulation of VEGF receptor expressions in human umbilical venous endothelial cells following VEGF stimulation. Hepatol Res 2001; 21: 261–7
- Morqillo F, Lee HY. Lonafarnib in cancer therapy. Expert Opin Investig Drugs 2006; 15: 709–19
- Satoh T, Endo M, Nakafuku M, Akiyama T, Yamamoto T, Kaziro. Accumulation of p21ras.GTP in response to stimulation with epidermal growth factor and oncogene products with tyrosine kinase activity. Proc Natl Acad Sci USA 1990; 87: 7926–9
- Gu WZ, Joseph I, Wang YC, Frost D, Sullivan GM, Wang L, et al. A highly potent and selective farnesyltransferase inhibitor ABT-100 in preclinical studies. Anticancer Drugs 2005; 16: 1059–69
- Oh SH, Kim WY, Kim JH, Younes MN, El-Naggar AK, Myers JN, et al. Identification of insulin-like growth factor binding protein-3 as a farnesyl transferase inhibitor SCH66336-induced negative regulator of angiogenesis in head and neck squamous cell carcinoma. Clin Cancer Res 2006; 12: 653–61
- Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, Somerfield MR. American Society of Clinical Oncology Technology Assessment: Chemotherapy sensitivity and resistance assays. J Clin Oncol 2004; 22: 3631–8