Abstract
Bluetongue viruses (BTVs) infect primarily domestic cattle and wild ruminants but have never been shown to infect normal human cells. Thus, humans are sero-negative towards BTVs. The selective and differential effects of BTV serotype 10 (BTV-10) infection were investigated with five cell lines including primary human embryo lung fibroblast (HEL) and primary murine embryos fibroblast(MEF), human hepatic carcinoma 3B cell line (Hep-3B), human lung carcinoma cell line (A549) and mouse fibroblast cell line (NIH 3T3). In this study, comparative analyses of differential cytopathic effects (CPEs), survival rates using different Multiplicities of Infection (MOI), ultra-structural changes by transmission electron microscopy, and the preferential cell cycle changes of infected cells by flow cytometry were made among these cells. Detection of the presence of BTV genome and kinetic analysis of virus titers in TCID50 were also made. We provided the first analytical demonstration and evidence that BTV-10 could selectively infect and degrade human cancer cells but not cultured primary normal cells. No CPE or viral mRNAs could be detected within these normal cells, while various degrees of CPE could be found in Hep-3B and A549, as well as in NIH 3T3 under similar conditions. Before death, BTV-infected human cancer cells were directly arrested in the sub-G1 phase and the diversity of BTV infection as shown by the MTT method had significant difference (F=95.635, p<0.01). Above results suggested that this viral dose-dependent cytotoxic effect is caused by both effective virion amplification and induced apoptosis. Cellular distinctive transformation status may contribute to the selectivity. Thus, selective degradation of human cancer cells but not normal diploid cells by the newly discovered oncolytic potential of BTV would provide a very attractive approach for cancer therapy in the future.
Bluetongue virus (BTV) is a non-enveloped, icosahedral virus and its genome contains ten segments of double-stranded RNA (ds-RNA). This arthropod-born virus is classified in the genus orbivirus within the Family Reoviridae. It infects domestic cattle, especially sheep with higher susceptibility and wild ruminants mainly through the biting midge, Culicoides. Infected livestock exhibit fever, an increased rate of respiration, nasal discharge with swelling, and cyanosis of the tongue. However, BTV has never been demonstrated to infect humans. Thus, humans are sero-negative against BTV. In vitro many cell lines are susceptible to BTV induced cytopathic effect (CPE) and virus-specific structures could be identified within infected cells later in infection, while at the same time some other cell lines are resistant to its infection Citation[1]. It is accepted that BTV infection of tissue culture cells depends upon a receptor-mediated endocytotic process and the BTV virion binds to the outer surface of susceptible cells via the larger of the two outer coat proteins, VP2, taken up via an endosomal route Citation[2], Citation[3]. But, until now, its exact molecular infectious mechanism still remains ambiguous.
In our recent studies, we have successfully cell culture-adapted a Bluetongue virus which isolated in Hubei, China and named BTV-HbC3. This unique BTV isolate could proliferate selectively and efficiently in both Hela Citation[4], Citation[5], Hep-3B and MA 782 cell Citation[6] and what's more, in SPC-A-1, BTV-HbC3 and BTV-10 induced similar cytopathology changes Citation[7], Citation[8]. BTV-HbC3 treated mice tumor model has also been established (unpublished data). All the results demonstrated that BTV-HbC3 can selectively infect the cancer cells and comparably did no harm to the normal tissue or cells. Since VP2, VP5 shows the most genomic and structural variation of all ten BTV proteins. Whether the standard serotype BTV-10 has this selective characteristic as well as BTV-HbC3 will give helpful sources for BTVs further relative oncolytic mechanism research.
In this study, we determined and compared the susceptibility of normal and human cancer cells to BTV-10 infection and explored whether oncolytic potential would be present in BTV similar to BTV-HbC3 and Reovirus. We demonstrated and showed for the very first time that BTV-10 did not infect normal human cells as we expected but could selectively degrade human cancer cells that showed differential susceptibility. Thus, BTVs could be used as attractive and selective virotherapy for human cancers in the future after in vivo and human clinical trials.
Materials and methods
Cells and viruses
Hep-3B (human hepatic cell carcinoma) Citation[9], A549 (human lung carcinoma) and NIH 3T3 (normal mouse fibroblasts) were obtained from the China Center for Type Culture (CCTCC), Wuhan, China. Hep-3B and NTH 3T3 cells were cultured and maintained in Dulbecco Modified Eagle's Medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum (FBS, China, SI JI QING) and 2 mM L-glutamine. A549 were grown in RPMI-1640 containing 10% FBS. The normal human embryonic lung (HEL) cells were obtained from a 93-day old aborted fetus, while MEF was obtained from the mouse embryo. All the cells were cultured as a monolayer culture according to the previous published protocol Citation[10], Citation[11] and maintained in a 37°C humidified incubator with 5% CO2.
Each of these five cells was separately infected with the BTV-10 within 24 h after the monolayer became confluent. BTV-10 was cell cultured–adapted by three consecutive passages in the Vero cell culture. The titer of the virus progeny had a 50% tissue culture infectious dose [TCID50] of 105.05 per ml and this virus stock was used in all sequent infections.
Cellular and morphological determination by light and electron microscopy
After cells were infected with BTV-10 at MOI of 1.0 for 36 h, the morphological changes of all the cells were observed and determined by light microscopy (LM). For EM preparation, cells were then collected, fixed in glutaraldehyde (30 g/L), and stained in osmium tetroxide (10 g/L) for 30 min at 4°C. It was dehydrated sequentially with acetone, embedded in EPON, and then polymerized at 60°C. The ultra-thin sections were stained with uranyl acetate and Reynold lead citrate before cellular ultrastructures were examined and determined by electron microscopy (EM; Hitachi H-600, Japan).
Detection of BTV genome by gel electrophoresis
Total RNA of the cells was extracted 36 h post infection by Trizol (GIBCO) according to the protocol provided by the manufacturer. The RNA pellet was then dissolved in 20 µl RNase-free water. Half of the 20 µl sample was used for electrophoresis on 1.5% agarose gel containing Ethidium bromide (EB) at constant voltage 5 V/cm.
Analysis of cell survival rate
Survival rate of the five cell lines was assessed by MTT assay which measured the cytotoxicity and cellular apoptosis by the bio-reduction of a tetrazoleum compound: 3-(4,5-dimethylthiazol-2-ly)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium into a formazan that could be measured colorimetrically. Each well of the 96 well plates was seeded with 4×104 cells and incubated with 200 µl of the growth media. After incubation for 24 – 48 h, cells were infected by BTV with MOI in series for 36 h. At the end of the incubation, the growth medium was removed and 20 µl MTT solution and 180 µl fresh medium was added to each well. Dimethyl sulfoxide (DMSO) was then added to each well and the optical density of each well was screened and determined with a Plate Reader (GENios VA200, TECAN, Australia) at 570 nm.
The survival rate=(optical density of the tested group ×100%)/the mean optical density of control. The titer of original viral suspension used was 105.05/ml TCID50. Based on the Poisson Distribution, TCID50=-In (0.5) = 0.693. Thus, 1 TCID50=0.693 IU (infection unit) Citation[10]. Through data conversion, different viral multiplicity of infection (MOI) in five groups was adapted to 10−3, 10−2, 10−1, 1 and 10 IU/cell, respectively.
In vitro viral replication assay
Hep-3B, A549 and NIH 3T3 cells were seeded into 24-well plates with a cell density adjusted to 1×105/well and with appropriate growth medium containing 2% FBS. The confluent cell monolayer was infected with BTV-10 at the MOI of 1.0. Time samples of mock and BTV-infected cells and medium were harvested at 6h, 12h, 24h, 36h and 48h pi. Three replicated wells were used for each time point and TCID50 was subsequently determined and titrated on Hep-3B cells.
To critically assess TCID50 of the virus progeny of each time point, the cell lysate and the medium of each time point were diluted 10-fold and 100 µl of each diluted sample was then inoculated onto monolayer of eight separate wells. After viral attachment, 400 µl fresh growth medium was added and the plates were incubated at 37°C with 5% CO2 and humidified air. The assay was terminated when cytopathic effects (CPE) were readily recognizable independently by technicians. The TCID50 dosage per milliliter was determined by using the formula of Karber.
TCID50 was calculated and determined by this equation: log TCID50=m = x a-D (S p-0.5) where xa is the last dilution index for which all of n cultures are infected (p = 1); D is the log of the dilution factor (log 10 = 1); Sp is the summation of p between the last dilution for which all of n cultures are infected (p = 1) and the first dilution for which all of n cultures are unaffected (p = 0).
Determination of the apoptosis rate and cell cycle arrest by propidium iodide staining and flow cytometry
Cells on monolayer and those in medium supernatant from the mock and BTV 10-infected groups were harvested, trypsinized, washed and resuspended in PBS. Cells were then fixed in 70% ethanol and stored overnight at 4°C. Cells were again pelleted and resuspended in 1 ml of PBS containing 0.2 mg/ml of propidium iodide (PI) and 0.15 g/ml of RNase A at 37°C or 4°C for 30 min in the dark. Stained cell samples were analyzed by flow cytometry (FCM; EPICS ALTRA II, Beckman) Citation[12]. Each deviated percentage of the two sub G1 phase got between mock and BTV-10 infected groups, was recognized as apoptosis index (AI). Analysis of DNA distribution was carried out with more than two independent experiments. All the values were determined, calculated and scored with more than 104 cells each time sample.
Statistical analysis
Comparisons among groups and inside the groups were statistically analyzed by ANOVA (Analysis of Variants). All data were analyzed with SAS 6.12 for Window.
Results
Cytopathic effects, morphological and ultrastructural changes of normal and human cancer cells infected by BTV-10
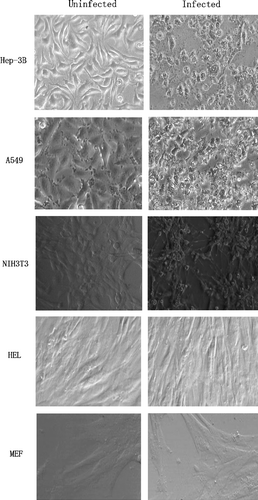
Over 90% cytopathic effects (CPE) were readily observed and detected in human cancer cells, Hep-3B and A549, infected with BTV 36h post-infection at a MOI of 1 as shown by light microscopy in . Cellular apoptosis was evident in these two human cancer cell lines. Morphological characteristics and CPE of BTV-infected NIH 3T3 were different. They had much less CPE but appeared to adhere to each other more tightly than Hep-3B and A549 cells before cell apoptosis occurred. While no visible CPE or pathological changes could be observed or detected in HEL and MEF cells even after 5th day post infection, they showed some minor morphological changes such as cell contraction with some loss of their normal spindle shape.
Figure 1. Light Microscopy of five cell lines infected by BTV-10 36 h post infection. Detection of morphological changes and CPE in BTV-10 infected two human cancer cell lines, (A549 and Hep-3B), the continuous cell line, NIH 3T3 and two primary fibroblast HEL and MEF. The magnification for all panels is ×200; except for MEF which is ×400.
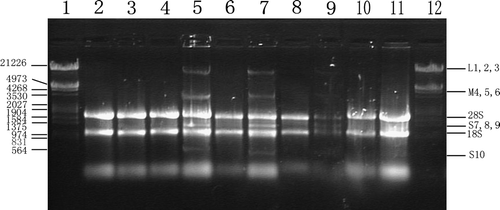
In electron microscopy (EM), BTV virions could be seen amplified efficiently in Hep-3B and NIH 3T3 cells, especially in A549 even released viral particles could be seen close to discontinuities in the plasma membrane (). In return, cellular CPE like dilated endoplasmic reticulum were readily detected. However, nucleic apoptosis characteristics like nuclear chromatin condensation and cytoplasmic shrinking were more easily observed in Hep-3B and NIH 3T3 than in A549, while most nucleus and mitochondria within A549 still remained morphologically unchanged. Most of all, compared with mentioned cell lines, HEL and MEF retained their normal cellular ultra structure 36 h pi and no virions could be found in the cytoplasm under the same conditions. In consistent with the above results, BTV viral genomic ds-RNA fragments could only be found in BTV-10 infected human cancer cells and NIH3T3 but not in normal HEL or MEF cells through gel electrophoresis as shown in .
Figure 2. Electron microscopy of five cell lines infected with BTV-10 36 h post infection. All of sections were stained with uranyl acetate and Reynold lead citrate. N stands for nucleus and the arrow shows the amplified virions. A. Hep-3B (×12 000), B. A549 (×20 000), C. NIH 3T3 (×15 000), D. HEL (×12 000) and E. MEF(×8 000).
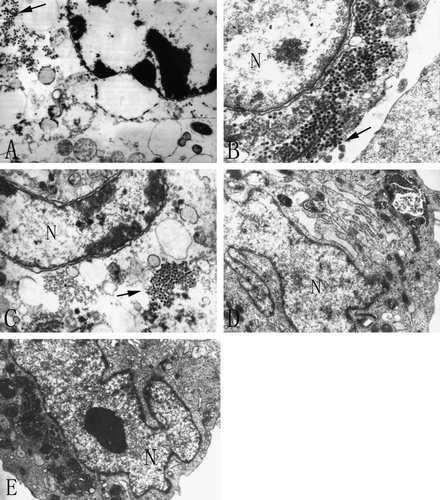
Figure 3. Gel electrophoresis of total RNA extract from different cell lines infected with BTV-10 36 h post infection. Lane 1. DNA Markers (λ DNA digested with Eco RI +Hind III); Lane 2. Total RNA extract from mock HEL cells; Lane 3. Total RNA isolated from BTV-10 infected HEL cells; Lane 4. Total RNA extract from mock A549 cells; Lane 5. Total RNA isolated from BTV-10 infected A549; Lane 6. Total RNA extract from mock Hep-3B cells; Lane 7. Total RNA isolated from BTV-10 infected Hep-3B; Lane 8. Total RNA extract from mock NIH3T3 cells; Lane 9. Total RNA isolated from BTV-10 infected NIH3T3; Lane 10. Total RNA extract from mock MEF cells; Lane 11. Total RNA isolated from BTV-10 infected MEF; Among all the lanes cellular ribosomal 28S and 18S rRNA could be seen, while only in 5, 7 and 9 lanes the three groups of BTV viral RNA bands (L M & S) could be detected.
Changes of survival rates in human cancer cells after BTV-10 infection
In our initial experiments, we used the MOI in series dilution to determine the survival rates of these five cell lines using the MTT assay. Statistic analysis by one-way ANOVA (Analysis of Variance) showed that there were significant differences among five different cell groups in their survival rates (F = 95.635, p < 0.01). There was also significant difference between every two groups by Multiple Comparisons, except between HEL and MEF cells (A).
Figure 4. Cytotoxic effect on the tested five cell lines after BTV-10 infection. (A) Multiple Comparisons show that difference between every two cell groups is significant (p** < 0.01), except difference between HEL and MEF cells (p* > 0.05). (B) Survival rate of Hep-3B, A549 and NIH3T3, after challenged by BTV-10, changes in accord with the series dilution of MOI. The data are the mean of eight wells.
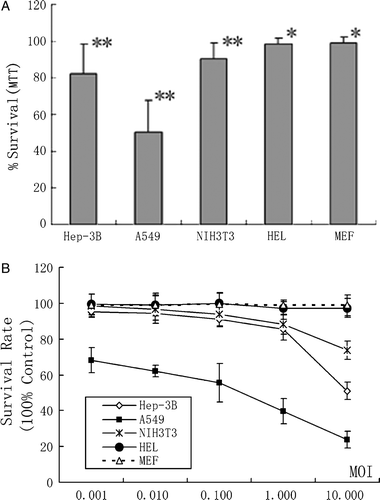
To further access whether survival rates of the human normal and cancer cells could be affected by different MOIs, we used series MOIs of BTV-10 to infect these five cell lines in multiple independent experiments and their survival rates were determined 36 h post infection by the MTT method. When the MOI of infection varied from 0.001 to 10, the survival rate of A549 cell declined gradually to about 20% 36 h p.i. (B). Its survival rate was reversibly proportional to the MOI used and always remained the lowest among the five cell lines tested in multiple determinations. BTV-10 infected Hep-3B cells could survive nicely with low dosage of MOI. However, when MOI was increased from 1 to 10, the survival rate of Hep-3B cells declined dramatically. This was similar for NIH 3T3 cells that could survive slightly better than Hep-3B cells when MOI was changed from 1 to 10. In contrast, the primary HEL and MEF cells survived very well up to 5 days independent of the MOI used for testing. These indicated that human cancer cells were preferentially degraded with higher MOI which could be important in subsequent in vivo and preclinical studies.
Human cancer cells infected with BTV-10 had more efficient viral replication and provided higher viral progeny yields
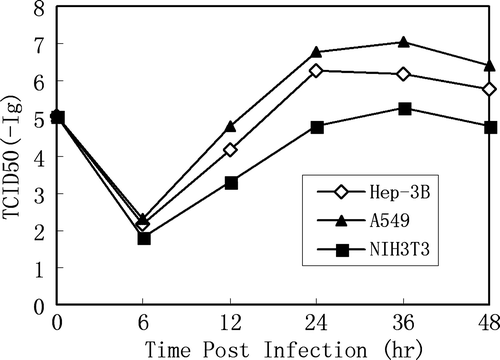
To assess replication efficiency of BTV in human cancer cells and their corresponding virus progeny yields, Hep-3B, A549 and NIH 3T3 cells were infected at a MOI of 1 and kinetic analysis of virus yields from different post infection time points was examined in virus infection assays (TCID50). HEL and MEF cells were included as negative control even though no viral progeny could be detected by this method.
Infected monolayer cells were scraped and pelleted. The cell pellets were resuspended in small volume of growth medium and cell lysates were prepared. TCID50 dosage per milliliter of each time point was then determined. As shown in , the low TCID50 at 6h pi represented the “uncoating” period and virus yield gradually reached a plateau between 24 – 36 h for both human cancer cell lines, A549 and Hep-3B. However, A549 provided higher viral titers among the three cell lines tested within the same time points pi in several independent experiments (). Cell apoptosis and death on these two human cancer cells 36 h and 48 h after infection was also visualized, determined and confirmed by crystal violet staining. In the case of NIH 3T3 fibroblast, comparatively less amount of infectious BTV-10 virions were obtained.
Figure 5. Virus yields of BTV-10 in various cell lines, including Hep-3B, A549 and NIH3T3. Monolayer cells were seeded on 24-well plates and infected with BTV-10 at MOI of 1. Virus yields were titered on Hep-3B cells after infection. Results represent the mean of triplicate wells.
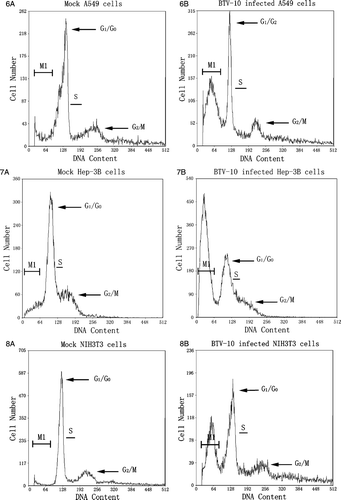
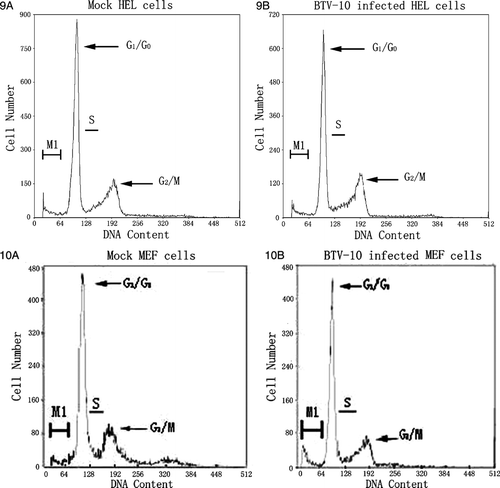
Selective cell cycle arrest in human cancer cells infected with BTV-10
During viral infections, virus actively interacts with the susceptible host cell. One of these active virus-host interactions is involved with the adjustment of host cell cycle regulation or cell cycle arrest. To examine whether cell cycle regulation was affected in human cancer cells after BTV 10 infection, cells were harvested 36 h p.i. at a MOI of 1, stained and evaluated by FCM. Result (–10) demonstrated BTV-10 only induced obvious cytotoxic effect in A549, Hep-3B, and NIH 3T3 cells, not in HEL and MEF. Among them A549 was the most susceptible cell line. When A549, Hep-3B, and NIH 3T3 cells were infected with BTV for 36 h, AI was significantly increased to 45.1%, 40.5% and 34.1% respectively. These represented significant differences in the sub-G1 peak between mock and BTV infected A549, Hep-3B, and NIH 3T3 cells. Besides, no obvious cell life cycle changes or arrest were found. This is different with Reovirus which can induce obvious G2/M cell cycle arrest in the absence of apoptosis Citation[13]. The induction and direct maintenance at sub-G1 peak by BTV infection or by other agents could be used to enhance the selective killing of human cancer cells in future.
Figure 6–10. Changes of DNA content within the cell cycle of five cell lines 36 h post-infection with BTV-10. M1 region in every figure stands for cells went into sub-G1 phase. Figure 6A is mock control of A549 and Figure 6B represents BTV-10 infected A549, AI counts for 45.1%. Figure 7A is mock control of Hep-3B and Figure 7B represents BTV-10 infected Hep-3B, AI counts for 40.5%; Figure 8A is mock control of NIH 3T3 and Figure 8B represents BTV-10 infected NIH 3T3, AI counts for 34.1%; Figures 9A,10A stand for mock control of HEL/MEF and Figures 9B,10B represent BTV infected HEL/MEF, there is no statistically significant difference within the changes of their AI which stands for 0.9% and 1.4% respectively.
Discussion
For the past century, no normal human cells have ever been successfully infected by BTV and thus, humans are normally sero-negative. In our study, two human cancer cell lines, (A549 and Hep-3B), two primary fibroblast HEL and MEF, and the immortalized mouse fibroblast cell line, NIH 3T3 were tested for their susceptibility to BTV infection. As shown by LM and EM ( and ), no CPE or morphological changes even virions could be found in HEL or MEF cells after BTV-10 infection. This resistant phenomenon is similar to previous studies in which Madin-Darby canine kidney (MDCK), Gekko lung and Rainbow trout gonad were proved resistant to BTV infection Citation[1], Citation[14]. Within these studies no viral antigens could be detected in cells even with immunological assays such as ELISA and RIP, which means the virion's amplification course could be totally blocked within the cytoplasm.
Pathological and morphological changes in virus infected cells are routinely determined by LM and EM. However, both LM and EM can reveal some morphological changes and both approaches are time-consuming and labor-intensive to determine these changes in large number of cells. Thus, they lack the statistical veracity. Similarly, DNA fragmentation method can only demonstrate/reveal a quasi-qualitative analysis of apoptotic cells Citation[12]. The percentage of apoptotic cells with destructive cellular membranes can not be compared and determined with quantitative accuracy. Thus, in this study, viral infection assays (TCID50), MTT method, flow cytometry counting and gel electrophoresis were also used in additional to LM and EM to provide more quantitative data for analysis. The combined results indicated that BTV-10 could replicate and amplify efficiently inside the cytoplasm of susceptible human cancer cells and amplification in abundance of the virus led to extensive CPE only to human cancer cells but not to normal human cells. Among them A549 was the most susceptible cell line. What's more, there is an interesting clue in this research that is, among the five cell lines we tested, both A549 and HEL have the human lung tissue origin, while both HEL, mouse MEF and mouse NIH3T3 belong to the fibroblast cell type. Since only HEL and MEF showed totally resistant to BTV-10's infection, it could be speculated that the different intracellular environment between diploid HEL and MEF cells and continuous A549 and NIH3T3 cell lines may be the possible cause led to BTV-10's selective infection.
The sigma 1 protein of Reovirus can induce G2/M cell cycle arrest in the absence of apoptosis Citation[13], Citation[15]. In contrast, BTV induced direct sub-G1 cell cycle arrest in BTV-infected human cancer cells before cell apoptosis (–10). Experiments are now in progress to determine whether and which BTV gene product(s) might be involved with this phenomenon in BTV infected human cancer cells.
Currently, there are many different mechanisms by which oncolytic viruses can selectively degrade cancer cells Citation[16–18]. However, when safety concerns are considered in potential human clinical trials and applications, dsRNA viruses such as BTV and Reovirus are more advantageous than the genetically modified DNA viruses since the integration of viral ss- and ds-RNA genomes into the host cell genome have not been shown or reported Citation[16], Citation[17]. Furthermore, the genomes of dsRNA viruses such as reovirus and BTV are much more stable than those of ss-RNA viruses that are more easily degraded. Since humans are 100% sero-positive to Reovirus Citation[15] but sero-negative to BTV, this suggests that the potential future use of BTV to degrade human cancer by direct intratumoral injection to degrade the injected tumors will not be inhibited by pre-existing immune components within the cancer cells. Thus, BTV is potentially better than Reovirus in oncolytic applications.
Both BTV and Reovirus are dsRNA viruses whose dsRNA genome fragments are good inducers of IFN. Thus, the induced IFN would activate a series signaling cascade involving multiple genes and pathways leading to the establishment of an ‘antiviral state’ such as PKR, 2′, 5′- oligo A synthetase, and Mx protein in normal cells Citation[19]. This might explain why normal human cells are resistant to BTV infection. However, PKR activation can be by blocked by Reovirus Sigma 3 protein and the “antiviral state” is blocked so that susceptible host cells can be infected by Reovirus. Even though the BTV protein(s) that might function as the Reovirus Sigma 3 protein have not been found yet, we hypothesize that such BTV protein does exist, the presence of which enable BTV to infect human cancer cells but not normal human cells that might have much stronger “antiviral state” which is more easily established.
The current mechanisms of oncolytic Reovirus towards human cancer cells have recently been summarized Citation[18]. An activated Ras/RalGEF/p38 pathway is potentially involved with the permissiveness of host cells to Reovirus infection Citation[20], Citation[21]. Since PKR phosphorylation and activity are impaired in most cancer cells and tumor, Reoviruses can successfully evade the cellular antiviral defense system, resulting in efficient viral amplification that leads to apoptosis of the infected host cell.
It has been shown that the c-K-ras gene inside the lung carcinoma cell line A549 that we used, has mutation at codon 12 Citation[22]. Since the Ras/RalGEF/p38 signal pathway inside the A549 cell has been activated, this might allow BTV to degrade human tumor cells efficiently similar to Reovirus. That is also why we detected more efficient BTV amplification in A549 cells than in Hep-3B cell () which is only a HBV-transformed cell line Citation[9]. Thus, we further hypothesize that the genetic alterations/mutations of different human cancer cells, such as signals within ras related pathway, play a key role in the susceptibility of different oncolytic viruses besides specific viral receptors which might be present in human cancer cells but not in normal human cells. Identification of BTV receptor(s) on the surface of the human cancer cells is also currently in progress.
In further comparative analysis of Reovirus and BTV, we have found that the normal mouse cell line NIH 3T3, is normally resistant to Reovirus infection, showed some degree of susceptibility to BTV infection. Since NIH3T3 is the cell line which has gain the ability for immortal passage in vitro, cellular genetic spontaneous transformation happens during passage course may contribute to this susceptibility. Therefore, compared with primary cultured MEF, within this circumstance we speculated that viral progeny then could amplify in amount in NIH3T3. What's more, since oncolytic Reovirus still shows cytotoxic effect to Madin-Darby canine kidney (MDCK) epithelial cells Citation[23], this intracellular environment difference existed by the cell type will be natural. But whether BTV-10 has this stricter selectivity than reovirus or not needs more evidence.
As to the cytotoxic effect mechanism, since both Reovirus and BTV are the members of Reoviridae and have dsRNA segments as their genomes, different gene products might also be involved with cell apoptosis of human cancer cells. The VP5 protein of BTV has recently been shown to induce or trigger apoptosis in mammalian cells Citation[24]. This is not surprising since this major outer capsid protein is glycosylated and can easily be inserted into the cellular membrane. Thus, we think that this protein might also be involved with the apoptosis of human cancer cells. Furthermore, the non-structural BTV protein, NS-3, has been shown to be a “viroporin”, the polymerization of which forms pores on the surface of BTV-infected cells, leading to the severe cellular leakage and potential cell death Citation[25]. We hypothesize that these two BTV proteins and other(s) are involved with the cytotoxic effect of human cancer cells after BTV-infection.
In general, since BTV-10's selective cytotoxic effect to the tumor cells was clearly indicated and demonstrated; further research about how to well manipulate this unique susceptibility will give us the theoretic bases to use it as an oncolytic agent for the safer application in human body in the future.
Acknowledgements
We thank Dr. Sanpu QU for providing three of cell lines used in this study, and Senior Lab Assistant Yan WANG, Xueju QU, and Senlin LEI for their excellent technical and computer assistance. This work was supported by National Natural Science Foundation of China (#30274471). Joseph K.-K. Li was supported in parts by Utah Agricultural Experiment Station Project 537.
Furthermore, the authors do not have any competing financial interest.
References
- Wechsler SJ, Mcholland LE. Susceptibilities of 14 cell lines to bluetongue virus infection. J Clin Microbiol 1988; 26: 2324–7
- Hassan S, Roy P. Expression and functional characterization of bluetongue virus VP2 protein: Role in cell entry. J Virol 1999; 73: 9832–42
- Mertens PPC, Diprose J. The bluetongue virus core: A nano-scale transcription machine. Virus Res 2004; 101: 29–43
- Dong CY, Chen DE, Chen X, Cao ZH, Duan JR, Liu YH. Isolation of bluetongue virus strain HbC and its biological characters. The Clinic Medicine Research Beijing. Chinese Population Publishing House, Beijing 1998; 180–182
- Chen X, Yan YF, Dong CY, Chen BP, Tu P, Wang FJ. The characteristics of the propagation, purification and serology of bluetongue virus HbC Strain. Acta Acad Med HuBei 1999; 20: 179–91
- Liang K, Dong CY, Zhang WY, Hu J, Chen J. The infectivity of bluetongue virus strain HbC3 to MA782 cell. Virologica Sinica 2006; 21: 168–71
- Xiao AT, Dong CY, Li JK-K, Chen DE, Liu J, Zhang WY. Studies on the infectivity of bluetongue virus strain HbC3 to several human and animal tumor cells. Virologica Sinica 2004; 19: 349–52
- Lei SL, Xiao AT, Liu CX, Chen BP, Fang P, Dong CY. Comparison of infectivity of BTV-HbC3 to SPC-A-1 with that of BTV-10 prototype. Med J Wuhan Univ 2004; 25: 669–71
- Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980; 209: 497–9
- Du P. Medical experimental virology. People's Military Medical Press, Beijing 1985; 102–109
- Xue QS. Principles and techniques of culture in vitro. Beijing Science Publishing House, Beijing 2001
- Kikuchi H, Ujiie S, Wakui A, Yokoyama A, Kanamaru R. Features of DNA oligonucleosomal fragmentation in human tumor cell lines and its detection by flow cytometry: Utility and limitation. Jpn J Cancer Res 1997; 88: 56–65
- Poggioli GJ, Keefer JL, Connolly TS, Dermody TS, Tyler KL. Reovirus-induced G2/M cell cycle arrest requires σ1s and occurs in the absence of apoptosis. J Virol 2000; 74: 9562–70
- McPhee DA, Parson IM, Della-Porta AJ. Comparative studies on the growth of Australian bluetongue virus serotypes in continuous cell lines and embryonated chicken eggs. Vet Microbiol 1982; 7: 401–10
- Norman KL, Lee PWK. Not all viruses are bad guys: The case for reovirus in cancer therapy. Drug Discov Today 2005; 10: 847–55
- Wildner O. Comparison of replication-selective, oncolytic viruses for the treatment of human cancers. Curr Opin Mol Ther 2003; 5: 351–61
- Vorburger SA, Pataer A, Swisher S, Gand Hunt KK. Genetically targeted cancer therapy: Tumor destruction by PKR activation. Am J Pharmacogenomics 2004; 4: 189–98
- Vidal L, Yap TA, White CL, Twigger K, Hingorani M, Agrawal V, et al. Reovirus and other oncolytic viruses for the targeted treatment of cancer. Target Oncol 2006; 1: 130–50
- Der SD, Zhou A, Williams B, Rand Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA 1998; 95: 15623–8
- Strong JE, Coffey MC, Tang D, Sabinin D, Lee PWK. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO 1998; 17: 3351–62
- Norman KL, Kirasawa K, Yang AD, Shields MA, Lee PWK. Reovirus oncolysis: The Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci USA 2004; 101: 11099–104
- Valenzuela DM, Groffen J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res 1986; 14: 843–52
- Rodgers SE, Barton ES, Oberhaus SM, Pike B, Gibson CA, Tyler KL, et al. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol 1997; 71: 2540–6
- Mortola E, Noad R, Roy P. Bluetongue virus outer capsid proteins are sufficient to trigger apoptosis in mammalian cells. J Virol 2004; 78: 2875–83
- Han ZY, Harty RN. The NS3 protein of Bluetongue virus exhibits viroporin-like properties. J Biol Chem 2004; 279: 43092–7