Abstract
Introduction. Erythropoiesis-stimulating agents (ESAs) are used in cancer therapy to reverse anaemia. It has been suggested that ESAs might improve treatment outcome by reducing tumour hypoxia, but ESAs might also increase tumour growth. In this work, the effect of recombinant human erythropoietin (rHuEpo) β was investigated on a human head and neck squamous carcinoma cell (HNSCC) line in vitro. The cell line was previously growth stimulated in combination with surgery in a xenograft model and the investigation was initiated to see if rHuEpo directly affects the tumour cell line, alone or in combination with cell stress, or if the in vivo effect should be attributed to secondary effects. Material and methods. The cell line LU-HNSCC-7 was grown in vitro and treated with rHuEpo alone or in combination with radiation, cisplatin, hypoxia or tumour extracts. The expression of the Epo receptor (EpoR) was investigated by western blotting after one- and two-dimensional electrophoresis, RT-PCR and through analysis of the effect on EpoR signalling. Results. The cell line was shown not to express EpoR. Furthermore, it was only possible to detect a minor effect on cell growth (1.4 times over control, p < 0.001) under specific conditions and at supra-pharmacological concentrations of rHuEpo β. No effect was detected on cell migration. None of the cell stressing treatments could enhance the minor growth stimulatory effect of rHuEpo β. Discussion. The conclusion is that rHuEpo β does not stimulate tumour growth of the investigated cell line through a direct interaction with tumour cells. We hypothesise that interactions with stromal cells and the stimulation of wound healing responses might, at least partly, explain the negative effects of ESA administration during cancer treatment. We propose that EpoR expression in HNSCC tumour cells might not be a good marker for prediction of ESA induced worsening of outcomes after cancer treatment.
Erythropoiesis-stimulating agents (ESAs), including the recombinant human erythropoietins (rHuEpo) epoetin alfa, epoetin beta and darbepoetin alfa, were originally used in cancer treatment to reverse chemotherapy-induced anaemia. Later, it has been suggested that ESAs might decrease tumour hypoxia and thereby improve the effectiveness of chemo- or radiotherapy Citation[1], Citation[2] but this has not been verified in clinical studies. Recently, however, the use of erythropoietin (Epo) in cancer treatment has been lively debated. Several clinical studies have shown decreased survival after Epo treatment as compared to placebo control. On the other hand, others have suggested that overall survival is improved Citation[3], Citation[4]. One explanation for these discrepancies that has been advocated is based on the differences in the target serum haemoglobin levels, being significantly higher in the studies showing negative effects of Epo. Too high haemoglobin levels might in fact, in addition to increasing the risk of thrombotic and vascular events, decrease the oxygenation of tumours by increasing the intra-capillary resistance and thereby affect the treatment outcome. The current recommendations are therefore to keep the haemoglobin level at or below 120 g/L Citation[4].
However, also other mechanisms for the negative effects of ESAs in cancer treatment have been suggested. Both erythropoietin and Epo receptor (EpoR) have been reported to be expressed in a large number of different primary tumours and tumour cell lines Citation[5] and it has been hypothesised that ESAs could stimulate growth and invasiveness of tumour cells. Experimentally, several such effects have been reported but the results are inconsistent. Thus, some groups have found Epo to increase tumour cell growth while others were unable to detect growth stimulation Citation[3], Citation[6]. In general, high concentrations (i.e. more than 10-fold higher than the serum concentrations achieved during Epo treatment) of Epo had to be used to detect significant increases in growth rate. Similarly, Moyheldin et al. Citation[7] and Lai et al. Citation[8] saw effects of Epo on the invasive properties of HNSCC cell lines in vitro while, for example, LaMontagne et al. Citation[9] could not detect any effects on migration properties of breast cancer cell lines.
Also, the detection of EpoR in cancer tissues and cells has been put under scrutiny. The detection of EpoR mRNA does not necessarily implicate the presence of active receptor, and the commercially available antibodies that have been used for immunohistochemistry detects several proteins unrelated to EpoR Citation[10]. In fact, an attempt to correlate the clinical finding of decreased locoregional progression-free survival in patients receiving Epo β to the expression of EpoR Citation[11] has been criticised because of the low specificity of the EpoR antibody used and the probable cross-reaction with Hsp70 Citation[12–15]. In vitro, the effects on intracellular EpoR signalling might be used to assess EpoR functionality. Thus, EpoR signalling has been described to be mediated by the consecutive phosphorylation of Janus kinase 2 (JAK2) and signal transducer and activator of transcription 5 (STAT5) Citation[16]. It has also been shown that Epo can activate mitogen-activated protein kinases (MAPKs) in the absence of JAK2/STAT5 phosphorylation in vascular smooth muscle cells Citation[17] and it has been shown that the effect of Epo in human melanoma cells is dependent on the phosphoinositide 3-kinase (PI3K)/Akt pathway.
Previously we have shown that an HNSCC cell line (that was xenografted to nude mice) was growth stimulated when exposed to a combination of rHuEpo β treatment and surgical transection, with the main stimulatory effect manifested during the first 4 days after the injury Citation[18]. Several other in vivo experiments have been reported but only in this work (the only one combining Epo with surgery) Epo treatment was shown to have a growth enhancing effect. In the present work, we have explored the effects of rHuEpo β on this cell line in vitro in order to see if it is possible to correlate the tumour growth acceleration with direct effects of rHuEpo β on the tumour cells. As the effect of rHuEpo β was detected only in surgically transected tumours, several types of cell stressing treatments, as well as other conditions presumably experienced by the tumour cells during injury, were combined with rHuEpo β in order to find conditions under which cell growth could be enhanced. Also, the effect on cell migration was investigated.
Materials and methods
Drugs and chemicals
Recombinant human erythropoietin β (NeoRecormon®) was obtained from Roche (Basel, Switzerland) and cisplatin (Cisplatin Ebewe) from Pharmalink AB (Upplands-Väsby, Sweden). All other reagents were of analytical grade.
Cell lines and growth conditions
The tumour line used, LU-HNSCC-7, was originally established from a recurrence of a moderately differentiated squamous cell carcinoma of the bucca (rT2 N0 M0). It was aneuploid in flow cytometry, with wild-type p53 (no mutation in exon 5-11) and without cyclin D1 amplification Citation[19]. The Epo-dependent human erythroleukemic cell line UT-7/Epo was a kind gift from prof. Patrick Mayeux (Institute Cochin de Génétique Moléculaire, Paris).
The LU-HNSCC-7 cells were maintained in logarithmic growth as monolayer culture in Dulbecco's modified Eagle's medium (D-MEM) from Invitrogen (Carlsbad, CA) supplemented with 10% heat-inactivated foetal calf serum (FCS) and antibiotics (100×103 units/L penicillin and 100×103 units/L streptomycin sulphate) under a humidified 5% CO2 atmosphere at 37°C. UT-7/Epo cells were grown in α-MEM (Invitrogen, Carlsbad, CA) with 10% FCS and 2×103 IU/l rHuEpo. To increase the expression of EpoR, the UT-7/Epo cells were grown for 24 h in Iscove's Modified Dulbecco's Medium (IMDM) with 0.4% BSA and 25 mg/L transferrin.
Cell lysis
Cells were lysed for 30 min at 4°C in RIPA buffer (1% Triton X-100, 0.1% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L 4-(2-aminoethyl)benzenesulphonyl fluoride (AEBSF), 2 mmol/L Na3VO4, 1 mmol/L NaF, 20 mmol/L Na4P2O7, 1 mmol/L EDTA and 50 mmol/L Tris-HCl, pH 7.4). Cell debris was removed by centrifugation at 18 600×g for 10 min. The protein concentrations of the lysates were determined with the Micro BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL) using bovine serum albumin as standard and the lysates were then stored at −80°C until use.
Western blotting
Cell lysates (15 µg protein per sample) were denatured in 0.5% SDS, 50 mmol/L dithiothreitol (DTT), 2.5% (V/V) glycerol, 0.13 mmol/L EDTA, 0.01% (W/V) bromophenol blue and 62 mmol/L Tris-HCl, pH 8.5 at 70°C for 10 min, and then separated either on 4–12% NuPAGE Bis-Tris gels with 3-(N-Morpholino)propanesulfonic acid (MOPS) buffer (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol or 7% SDS-PAGE gels with 0.12% SDS, 0.38 mol/L glycine and 49 mmol/L Tris, pH 8.8 as running buffer. In the two-dimensional electrophoresis the samples were first separated in a 7% SDS-PAGE gel. Each lane was then cut out and transferred to 4–12% NuPAGE Bis-Tris gels and separated in MOPS buffer.
After electrophoresis, the proteins were transferred to PVDF-membrane in 192 mmol/L glycine and 25 mmol/L Tris, pH 8.3. Immunoreactivity was probed with the EpoR antibodies H-194 (N-terminal) and C-20 (C-terminal) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-STAT5 (Tyr694), anti-phospho-Akt (Ser473) and anti-phospho-p44/p42 MAP kinase (Thr202/Tyr204) from Cell Signaling Technology (Danvers, MA), anti-phospho-JAK2 (Tyr1007/1008) from Upstate (Lake Placid, NY). As loading control, anti-actin (H-196) from Santa Cruz Biotechnology or anti-pan-actin from Cell Signaling Techology was used. The primary antibody was detected with an anti-rabbit IgG horseradish peroxidase-linked F(ab')2 fragment from donkey in combination with ECL-plus detection reagent (Amersham Biosciences, Little Chalfont, UK).
RT-PCR
Total RNA was prepared from cell pellets (separately from three different batches for each combination of cell line and growth condition) using the PureLink Micro-to-Midi total RNA purification system from Invitrogen. The cDNA and PCR reactions were performed in triplicate with 200 ng RNA in 10 µL per reaction the SuperScript III Platinum SYBR green one-step qRT-PCR kit (Invitrogen) in a Rotor-Gene RG-3000 (Corbett Research, St. Neots, UK) with program 50°C for 3 min, 95°C for 5 min, and 40 cycles with 95°C for 15 s and 60°C for 30 s. Primer sequences for EpoR were forward 5′-GCTTCCCTGGAAGTCCTCTC-3′ and reverse 5′-CTATGTCCACACTGCCACCA-3′, and for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward 5′-GAGTCAACGGATTTGGTCGT-3′ and reverse 5′-TGGAAGATGGTGATGGGATT-3′. All oligonucleotides were from DNA Technology (Risskov, Denmark). After completion of the PCR reaction, the products were separated by polyacrylamide gel electrophoresis and visualised by silver staining.
rHuEpo incubations
For cell growth measurements, 5×105 LU-HNSCC-7 cells in D-MEM with 10% FCS were seeded into 60 mm dishes in at least triplicate. The medium was switched to serum free medium with the indicated additions of rHuEpo 24 h after seeding. After seven days of rHuEpo treatment, the cells were harvested and viable cells counted in a cell counting chamber using 0.4% trypan blue solution in phosphate buffered saline (PBS).
For examination of EpoR downstream phosphorylation the UT-7/Epo cells were starved in IMDM with 0.4% BSA and 25 mg/L transferrin and the LU-HNSCC-7 cells in D-MEM without FCS for 24 h. rHuEpo was then added in the corresponding media. The stimulation was stopped by two PBS washes and the cells were then lysed.
Combination of cell stress and rHuEpo β
Irradiation
Before irradiation, the cells were treated for 72 h with 0 to 50×103 IU/L rHuEpo in D-MEM supplemented with 10% FCS. After this treatment, the cells were harvested and suspended in medium with rHuEpo in 15 mL tubes. The tubes were irradiated with single doses between 0 to 5 Gy in a Gamma cell 2000 (Mølsgaard Medical, Denmark). Within one hour after harvest the cells were seeded in 96-well plates (2 000 cells/well) and then incubated for seven days. Cell survival was estimated by the sulforhodamine B (SRB) assay as described below.
Cisplatin treatment
Cells were seeded in 100 µL D-MEM with 10% FCS and 0 to 10×103 IU/L rHuEpo into 96 well-plates (103 cells/well) 48 h before initiating the treatment. Cisplatin was added (final concentrations 0 to 50 mmol/L) and incubated with the cells for one hour. Thereafter, the drug-containing medium was removed, and the cells were allowed to regrow in D-MEM with FCS and rHuEpo as before for five days. Cell numbers were estimated by the SRB assay.
Sulforhodamine B assay
To stop cell growth, 25 µL cold (8°C) 50% trichloroacetic acid was added to each well, and the plates were incubated for 60 min at 8°C. The supernatant was discarded and the plates were washed five times with tap water and air-dried. The fixed cells were stained by the addition of 25 µL SRB solution (0.4% [W/V] SRB in 1% acetic acid) per well and incubated for 10 min at room temperature. After staining, the plates were rinsed five times with 1% acetic acid to remove unbound SRB, and again allowed to air-dry. The dye was dissolved in 100 µL 10 mmol/L Tris base and the absorbance at 570 nm was measured in a Multiscan MS (Labsystems, Finland).
Hypoxia
Exposure to hypoxic conditions was performed in an enclosed chamber flushed with a premixed gas mixture (5% CO2, 95% N2). The O2-concentration was maintained at 1% or less throughout the course of the experiment. Cells were seeded into 60 mm dishes (5×105 cells/dish) with or without rHuEpo (10×103 IU/L). After 24 hours, the dishes were entered into the enclosed chamber for 4, 8 or 12 h and then incubated under ordinary conditions. Cells were counted 5 days after seeding.
Tumour extracts
Extracts were obtained from solid tumours of cells grown as xenografts on nude mice in order to examine the effect of trauma derived factors on tumour cell growth. The experiment was approved by the local ethics committee of Lund University (M 82-03). Twenty-three days after transplantation the tumours were excised, weighed and chilled on ice. They were cut into small pieces with a scalpel blade, and then transferred to a Dounce glass homogenizer. Four volumes of ice-cold PBS with 0.2 mmol/L AEBSF and 0.2 mmol/L EDTA were added and the tissue homogenised by 20 up-and-down strokes with a loose fitting pestle. Particulate matter was removed by centrifugation at 18 600×g for 30 min followed by filtration through a 0.2 µm sterile filter. The protein concentration of the extracts was determined with the Micro BCA Protein Assay Kit with bovine serum albumin as standard. The extracts were stored at −80°C until use. To measure the effect of tumour extract on tumour cell growth, 104 cells were seeded into at least ten 35 mm dishes in D-MEM with 10% FCS with or without tumour extract (protein concentration 10 mg/L) and rHuEpo (10×103 IU/L). Seven days after seeding, cells were trypsinised and harvested. Viable cells were counted in a NucleoCounter (ChemoMetec A/S, Allerød, Denmark).
Migration assay
In vitro migration of tumour-cell was studied in Boyden chambers (Neuro Probe Inc, Gaithersburg, MD) using polyvinylpyrrolidone-free polycarbonate filters with 8 µm pore size coated with fibronectin. Cells suspended in D-MEM supplemented with 0.1% bovine serum albumin (BSA) were seeded into the upper chamber, and were allowed to migrate against different concentrations of rHuEpo or tumour extract in D-MEM with 0.1% BSA in the lower chamber.
The chambers were incubated at 37°C with 5% CO2 for 16 h. After the incubation, the membranes were fixed in 99.5% methanol for 15 min, and stained with hematoxylin. The cells on the upper surface of the membrane were completely removed with a cotton swab and those on the lower side were counted under a light-microscope in three different fields for every membrane.
Results
The Epo receptor
LU-HNSCC-7 was earlier probed for EpoR expression using the EpoR antibody C-20 in immunohistochemistry Citation[18]. Lately, the use of this antibody in immunohistochemistry has been questioned Citation[10], which led us to further investigate the expression of EpoR using western blotting with the Epo dependent cell line UT-7/Epo as a positive control.
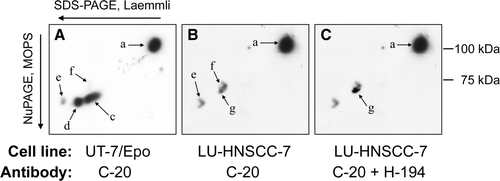
The C-20 antibody bound to five different proteins (a-e) in the UT-7/Epo lysate, separated in a 7% SDS-PAGE gel according to Laemmli (). Three of these bands (band b, 73 kDa, c, 69 kDa and d, 64 kDa) have earlier been described to be different forms of EpoR Citation[20] and they also agreed in molecular weight to what has been described by Epo cross-linking experiments Citation[21], Citation[22]. In accordance with the results of Verdier et al. Citation[20], band c was down-regulated after long-term exposure to rHuEpo β (lane 1 and 4) while band b was visible only after short-term stimulation with rHuEpo β (lane 3). The LU-HNSCC-7 lysate contained band a (116 kDa) and e (58 kDa), the origin of which has not been identified, but also two bands (f and g) at approximately 69 kDa (lane 5). Band g was also detected by the EpoR antibody H-194 (this reactivity was also present in some UT-7/Epo lysates but at much lower concentration [not shown]).
Figure 1. Cell lysates were separated by electrophoresis: lane 1–6 in a 7% SDS-PAGE gel according to Laemmli, and lane 7–10 in a 4–12% NuPAGE, Bis-Tris gel with MOPS buffer. Lanes 1–5 and 7–9 were probed with anti-EpoR C-20 and lanes 6 and 10 with H-194. UT-7/Epo cells were grown with 2×103 IU/L rHuEpo β (lanes 1 and 7) or without rHuEpo β for 24 h (lanes 2 and 8). Alternatively, they were stimulated for 10 min (lane 3) or 60 min (lane 4) with 10×103 IU/L rHuEpo β. LU-HNSCC-7 cells were grown with 10% FCS (lanes 5, 6, 9 and 10). Band b, c and d have earlier been identified in UT-7/Epo as different forms of EpoR while band a and e were unrelated to EpoR Citation[20]. Band f and g were present in LU-HNSCC-7.
![Figure 1. Cell lysates were separated by electrophoresis: lane 1–6 in a 7% SDS-PAGE gel according to Laemmli, and lane 7–10 in a 4–12% NuPAGE, Bis-Tris gel with MOPS buffer. Lanes 1–5 and 7–9 were probed with anti-EpoR C-20 and lanes 6 and 10 with H-194. UT-7/Epo cells were grown with 2×103 IU/L rHuEpo β (lanes 1 and 7) or without rHuEpo β for 24 h (lanes 2 and 8). Alternatively, they were stimulated for 10 min (lane 3) or 60 min (lane 4) with 10×103 IU/L rHuEpo β. LU-HNSCC-7 cells were grown with 10% FCS (lanes 5, 6, 9 and 10). Band b, c and d have earlier been identified in UT-7/Epo as different forms of EpoR while band a and e were unrelated to EpoR Citation[20]. Band f and g were present in LU-HNSCC-7.](/cms/asset/604be537-cfb2-4688-96f0-b8ce70523f05/ionc_a_391527_f0001_b.gif)
As band d, which is a maturing form of EpoR, was missing in the LU-HNSCC-7 cell line, the possibility that either band f or g could be identical to band c in the UT-7/Epo cells seemed questionable but not possible to resolve by this technique alone. Interestingly, the migration of several of the bands was shifted when the same cell lysates were separated in a 4–12% NuPAGE Bis-Tris gel with MOPS buffer () which enabled us to perform a two-dimensional electrophoresis using 7% SDS-PAGE in the first dimension and 4–12% NuPAGE with MOPS buffer in the second dimension (). With this technique, it was clear that the classical EpoR, represented by spot c and d (, panel A), was not present in the LU-HNSCC-7 cells (, panels B and C).
Figure 2. Separation of anti-EpoR reactive proteins by two-dimensional electrophoresis. First dimension: SDS-PAGE according to Laemmli, second dimension: 4–12% NuPAGE, Bis-Tris with MOPS buffer. A. Epo starved UT-7/Epo cells probed with C-20. B. Serum starved LU-HNSCC-7 cells probed with C-20. C. LU-HNSCC-7 cells probed first with C-20 and then, after stripping, with H-194. The images were overlaid electronically and matched using the membrane outlines. The background in the H-194 image was subtracted so that only the immunoreactive spot was visible (arrow g). Spot a and c-g correspond to band a and c-g in , i.e. spot c and d represent different forms of EpoR. Spot f and g, present mainly in LU-HNSCC-7, were separate from the EpoR spots.
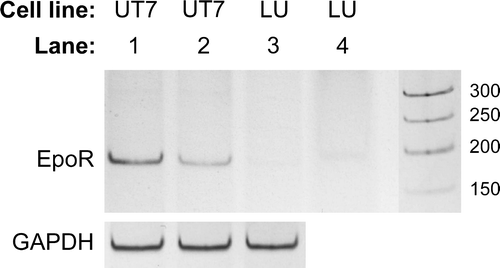
To further establish if the EPOR gene was expressed by LU-HNSCC-7 cells RT-PCR was performed on cell extracts, again using UT-7/Epo cells grown with or without rHuEpo β as positive control (). As evident, there was no detectable expression of EpoR mRNA in the LU-HNSCC-7 cell line while UT-7/Epo grown with Epo expressed EpoR mRNA that was up-regulated when the cells were deprived of Epo for 24 h.
Figure 3. RT-PCR of EpoR mRNA. PCR products from all (3×3) replicates of each combination of cell line and growth condition were pooled and separated by polyacrylamide gel electrophoresis. UT-7/Epo cells grown without Epo for 24 h (lane 1), or with 2×103 IU/L rHuEpo (lane 2), and LU-HNSCC-7 cells (lane 3). In lane 4, 10-fold higher amount of the LU-HNSCC-7 product was loaded. GAPDH mRNA was probed for relative quantification.
EpoR signalling pathway
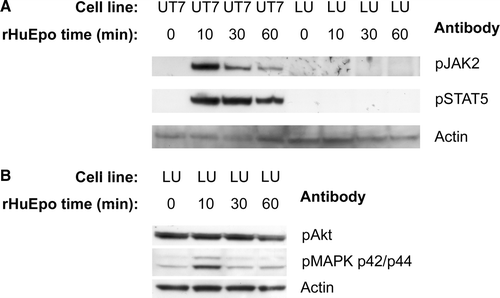
In order to see if any of the described EpoR coupled intracellular signalling pathways were activated by incubation with Epo, the phosphorylation of JAK2, STAT5, p44/p42 MAPK and Akt was probed with antibodies against the phosphorylated species. In the control cell line, UT-7/Epo Citation[23], there was a fast phosphorylation of JAK2, peaking at 10 min, followed by a somewhat delayed phosphorylation of STAT5. This was in sharp contrast to the LU-HNSCC-7 cell line where there was no detectable phosphorylation of JAK2 or STAT5 (A). Akt was constitutively phosphorylated in the LU-HNSCC-7 cell line and there was no measurable increase in the phosphorylation after rHuEpo β stimulation. The phosphorylation of p44/p42 MAPK, however, was somewhat increased after 10 min but returned to baseline after 30 min (B).
Figure 4. Cell lysates from UT-7/Epo and LU-HNSCC-7 cells stimulated with 10×103 IU/L rHuEpo β for the indicated times were separated by electrophoresis, transferred to PVDF membrane and probed consecutively with anti-phosho-JAK2, anti-phospho-STAT5 and anti-actin (A) or anti-phospho-Akt, anti-phospho-p44/p42 MAPK and anti pan-actin (B).
Effect of rHuEpo β
To investigate if rHuEpo β can stimulate tumour cell growth per se or in combination with cell or tissue injury associated factors, the LU-HNSCC-7 cells were grown in vitro and treated under a wide range of different conditions. The exact conditions used for the incubations with rHuEpo were chosen based on earlier experiences with other substances and on theoretical assumptions of possible effects of Epo in combination with the different cell stressing agents. There were no consistent effects of rHuEpo β on tumour cell growth under ordinary cultivation conditions with 10% heat inactivated FCS in the medium or when the cells were starved 24 h prior to stimulation and then treated with rHuEpo β in the absence of FCS (data not shown). Similarly, the combination of cell injury with radiation up to 5 Gy or cisplatin up to 50 mmol/L for one hour, or the exposure to hypoxic conditions or tumour extracts did not result in any measurable effect of rHuEpo β (data not shown).
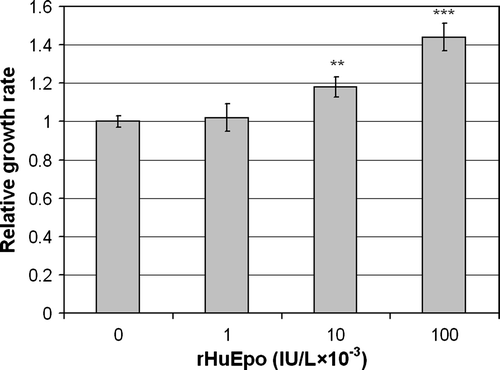
Only when the cells were treated with rHuEpo β in serum free medium but without pre-starvation there was a dose dependent effect on cell growth with significant increases of 1.2 times with 10×103 IU/L rHuEpo β (p = 0.008) and 1.4 times with 100×103 IU/L (p = 10−5) (). The growth was, however, much lower than in serum containing medium (not shown).
Figure 5. Effect of rHuEpo β on serum-deprived LU-HNSCC-7 cells. Cells were seeded in 60 mm dishes. Twenty-four hours later, the cells received new medium without serum but with the indicated amounts of rHuEpo β and were incubated for seven days before cell counting. The results were calculated as the average of four independent experiments with triplicate measurements. Error bars indicate standard error of the mean (SEM) (**p < 0.01 and ***p < 0.001).
We also analyzed the effect of rHuEpo β on the migration of the LU-HNSCC-7 cell line using Boyden chambers. In this set-up there was no significant increase of migration with the addition of rHuEpo β or a combination of rHuEpo β and the cell-free tumour extract (data not shown).
Discussion
Several clinical studies have indicated that adjustment of blood haemoglobin concentrations with ESAs during cancer therapy might adversely affect treatment outcome and lead to decreased survival compared to patients receiving placebo. It has been suggested that target haemoglobin concentrations higher than 120 g/L in the studies showing lower survival with ESA treatment might explain the outcome. However, there have also been indications that ESAs could directly stimulate tumour growth or malignancy and a thorough knowledge of the molecular mechanisms behind the negative effects of ESAs might help us to improve treatment schedules and also to increase the utility of the compounds.
In the present work we were not able to detect any direct effects of rHuEpo β in vitro that could readily explain the growth stimulation earlier seen in vivo in combination with surgery for the same cell line Citation[18]. Neither could the hypothesis that cellular stress might induce Epo responsiveness be verified. The only effect of rHuEpo β was recorded at supra-pharmacological concentrations and under very specific conditions (similar to effects earlier described for other HNSCC cell lines Citation[8]). Thus, the cells had to be cultivated for 7 days in the absence of serum additive with more than 10×103 IU/L rHuEpo β for a low increase in growth over control (≤1.4 times) to be significantly manifested. Furthermore, pre-starving the cells for 24 h prior to rHuEpo β exposure abolished the response. Possibly, one or more co-stimulatory factors present in FCS remained in the medium at sufficient concentrations when the medium was changed without pre-starvation. Hypothetically, if the conditions used were sub-optimal for Epo stimulation, other (more optimal) conditions might support tumour growth stimulation by pharmacological concentrations of rHuEpo β. However, the present work did not suggest that this could be the case as none of the tested manipulations led to stimulation by lower rHuEpo β concentrations. Therefore, it is highly unlikely that this single effect could explain either the previously reported stimulation of xenografted tumours or the clinically described adverse effects of ESA treatment.
The cell line that was used in this work, LU-HNSCC-7, did not express the Epo receptor as shown by a combination of western blotting, RT-PCR and measurement of JAK2/STAT5 phosphorylation at Epo exposure. Thus, the recorded effects of rHuEpo β, including p44/p42 MAPK phosphorylation and the above mentioned growth stimulation, were not due to EpoR signalling but must have been dependent on one or several other receptors. Taken together, the lack of EpoR expression in LU-HNSCC-7 and the minor effects of Epo on cell growth in vitro indicate that the previously recorded Epo induced tumour growth increase Citation[18] was not maintained through a direct effect of Epo on the cancer cells. Instead, the possibility that Epo interacts with other cells present in the tumour stroma seems more attractive. In favour of this, it has been described that Epo can affect wound healing responses, possibly through the interaction with macrophages, endothelial cells or via stimulation of vascular endothelial growth factor (VEGF) secretion Citation[24], Citation[25]. During cancer treatment, this might lead not only to improved tissue healing after tumour removal, but also to stimulation of the growth of remaining viable tumour cells and, thereby, to decreased loco-regional control and overall survival.
Of course, Epo might affect other HNSCC cell lines in different ways, for example by stimulating invasiveness via EpoR as reported by Lai et al., or the combination of direct and indirect effects might cooperatively enhance the impact of Epo treatment. In any case, our results indicate that EpoR expression in tumour cells might not solely predict the response of tumours to ESA treatment.
The use of antibodies for the detection of EpoR in histological sections and after western blotting has been widely debated. We showed that the C20 antibody indeed binds to EpoR but also to other proteins with similar apparent molecular masses. This precludes the uncritical use of the antibody for detection of EpoR after ordinary electrophoresis and blotting, and definitely for the detection of EpoR by immunohistochemistry. The 2D electrophoresis technique developed in this work could be used (in combination with appropriate positive controls) to assess the expression of active EpoR in tumour cell lines. However, to correctly discern tumour versus stroma cell expression of EpoR in tumour tissue the development of specific antibodies detecting active EpoR is still a critical issue.
In conclusion, stimulation of HNSCC tumour growth by rHuEpo β might not necessarily be coupled to the expression of EpoR in the tumour cells. We hypothesise that interactions with stromal cells and the stimulation of wound healing responses might, at least partly, explain the negative effects of ESA administration during cancer treatment and therefore we propose that other markers than EpoR expression in HNSCC tumour cells should be explored to predict negative effects of ESAs during cancer treatment.
Acknowledgements
This investigation was supported by the Swedish Cancer Society (1304-B04-18XAC and 4839-B04-02PBC), the King Gustaf V Jubilee fund (04-4251), governmental funding of clinical research within the NHS, Region of Scania R&D funding, the Foundations of the University Hospital of Lund, and the Foundation Laryngfonden. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Henke M, Laszig R, Rübe C, Schäfer U, Haase K-D, Schilcher B, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: Randomised, double-blind, placebo-controlled trial. Lancet 2003; 362: 1255–60
- Leyland-Jones B, Semiglazov V, Pawlicki M, Pienkowski T, Tjulandin S, Manikhas G, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: A survival study. J Clin Oncol 2005; 23: 5960–72
- Jelkmann W, Bohlius J, Hallek M, Sytkowski AJ. The erythropoietin receptor in normal and cancer tissues. Crit Rev Oncol Hematol 2008; 67: 39–61
- Melosky BL. Erythropoiesis-stimulating agents: Benefits and risks in supportive care of cancer. Curr Oncol 2008; 15: S10–S15
- Hardee ME, Arcasoy MO, Blackwell KL, Kirkpatrick JP, Dewhirst MW. Erythropoietin biology in cancer. Clin Cancer Res 2006; 12: 332–9
- Sinclair AM, Todd MD, Forsythe K, Knox SJ, Elliott S, Begley CG. Expression and function of erythropoietin receptors in tumors: Implications for the use of erythropoiesis-stimulating agents in cancer patients. Cancer 2007; 110: 477–88
- Mohyeldin A, Lu H, Dalgard C, Lai SY, Cohen N, Acs G, et al. Erythropoietin signaling promotes invasiveness of human head and neck squamous cell carcinoma. Neoplasia 2005; 7: 537–43
- Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A, et al. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene 2005; 24: 4442–9
- LaMontagne KR, Butler J, Marshall DJ, Tullai J, Gechtman Ze, Hall C, et al. Recombinant epoetins do not stimulate tumor growth in erythropoietin receptor-positive breast carcinoma models. Mol Cancer Ther 2006; 5: 347–55
- Elliott S, Busse L, Bass MB, Lu H, Sarosi I, Sinclair AM, et al. Anti-Epo receptor antibodies do not predict Epo receptor expression. Blood 2006; 107: 1892–5
- Henke M, Mattern D, Pepe M, Bezay C, Weissenberger C, Werner M, et al. Do erythropoietin receptors on cancer cells explain unexpected clinical findings?. J Clin Oncol 2006; 24: 4708–13
- Agarwal N, Gordeuk VR, Prchal JT. Are erythropoietin receptors expressed in tumors? Facts and fiction–more careful studies are needed. J Clin Oncol 2007; 25: 1813–4
- Della Ragione F, Cucciolla V, Borriello A, Oliva A, Perrotta S. Erythropoietin receptors on cancer cells: A still open question. J Clin Oncol 2007; 25: 1812–3
- Jelkmann W, Laugsch M. Problems in identifying functional erythropoietin receptors in cancer tissue. J Clin Oncol 2007; 25: 1627–8
- Sizer KC. Heat shock protein 70, erythropoietin, and cancer. J Clin Oncol 2007; 25: 4326
- Wojchowski DM, Gregory RC, Miller CP, Pandit AK, Pircher TJ. Signal transduction in the erythropoietin receptor system. Exp Cell Res 1999; 253: 143–56
- Ammarguellat F, Llovera M, Kelly PA, Goffin V. Low doses of EPO activate MAP kinases but not JAK2-STAT5 in rat vascular smooth muscle cells. Biochem Biophys Res Commun 2001; 284: 1031–8
- Kjellén E, Sasaki Y, Kjellström J, Zackrisson B, Wennerberg J. Recombinant erythropoietin beta enhances growth of xenografted human squamous cell carcinoma of the head and neck after surgical trauma. Acta Otolaryngol 2006; 126: 545–7
- Henriksson E, Baldetorp B, Borg A, Kjellen E, Akervall J, Wennerberg J, et al. p53 mutation and cyclin D1 amplification correlate with cisplatin sensitivity in xenografted human squamous cell carcinomas from head and neck. Acta Oncol 2006; 45: 300–5
- Verdier F, Gomez S, Lacombe C, Mayeux P. Selected anti-Epo receptor antibodies predict Epo receptor expression. Blood 2006; 108: 1106
- Jones SS, D'Andrea AD, Haines LL, Wong GG. Human erythropoietin receptor: Cloning, expression, and biologic characterization. Blood 1990; 76: 31–5
- Wognum AW, Lansdorp PM, Humphries RK, Krystal G. Detection and isolation of the erythropoietin receptor using biotinylated erythropoietin. Blood 1990; 76: 697–705
- Verdier F, Walrafen P, Hubert N, Chrétien S, Gisselbrecht S, Lacombe C, et al. Proteasomes regulate the duration of erythropoietin receptor activation by controlling down-regulation of cell surface receptors. J Biol Chem 2000; 275: 18375–81
- Buemi M, Galeano M, Sturiale A, Ientile R, Crisafulli C, Parisi A, et al. Recombinant human erythropoietin stimulates angiogenesis and healing of ischemic skin wounds. Shock 2004; 22: 169–73
- Haroon ZA, Amin K, Jiang X, Arcasoy MO. A novel role for erythropoietin during fibrin-induced wound-healing response. Am J Pathol 2003; 163: 993–1000