Abstract
Sequencing of the human genome has recently been completed and mapping of the complete genomic variation is ongoing. During the last decade there has been a huge expansion of studies of genetic variants, both with respect to association studies of disease risk and for studies of genetic factors of prognosis and treatments response, i.e., pharmacogenomics. The use of genetics to predict a patient's risk of disease or treatment response is one step toward an improved personalised prevention and screening modality for the prevention of cancer and treatment selection. The technology and statistical methods for completing whole genome tagging of variants and genome wide association studies has developed rapidly over the last decade. After identifying the genetic loci with the strongest, statistical associations with disease risk, future studies will need to further characterise the genotype-phenotype relationship to provide a biological basis for prevention and treatment decisions according to genetic profile. This review discusses some of the general issues and problems of study design; we also discuss challenges in conducting valid association studies in rare cancers such as paediatric brain tumours, where there is support for genetic susceptibility but difficulties in assembling large sample sizes. The clinical interpretation and implementation of genetic association studies with respect to disease risk and treatment is not yet well defined and remains an important area of future research.
Human genetic variations – polymorphisms
Single nucleotide polymorphisms (SNP), where a single nucleotide (adenine (A), cytosine (C), thymine (T), guanine (G)) is changed, is the most common type of human genetic variant. The general definition of a SNP is that the rare homozygote variant has a frequency of at least 1%. It is currently estimated that there are 11 million SNPs in the human genome and about seven million of these have a frequency of 5% or higher (see HapMap project www.hapmap.org) Citation[1]. SNPs that change the amino acids in the protein and thereby the potential protein functions are called non-synonymous SNPs. Groups of SNPs are usually inherited together in chromosomal regions described as haplotype blocks. These blocks may span over small or large segments of a gene, depending on the degree of genetic variability. Differences in haplotype block structure across genes are exemplified by the epidermal growth receptor family EGFR (HER1) and ERBB2 (HER2), two genes within the same receptor family but with very different genetic variability (). SNPs in the same haplotype block are usually inherited together, therefore “tagging SNPs” can be chosen in place of measuring all SNPs in the haplotype block. Genetic variants that are strongly correlated are also described as being in linkage disequilibrium (LD) with each other (). Using this knowledge, the haplotype mapping HapMap project has been able to identify 500 000 genomic variants that explains about 80% of the human genomic variation, through the mechanism of haplotype tagging and correlation of tagging SNPs to the other polymorphisms Citation[1], Citation[2]. More recently, commercial chips have been developed for genome wide association studies using tagging SNPs that enable rapid genotyping and analyses of many cases.
Figure 1. Linkage disequilibrium (LD) haplotype blocks of the EGFR gene (A) and ErbB2 gene (B). Exons have been redrawn to show the relative positions in the genes, therefore maps are not to physical scale.
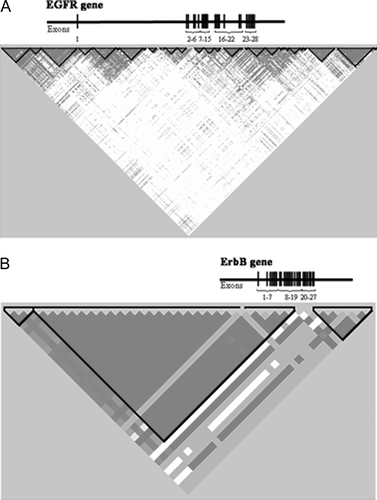
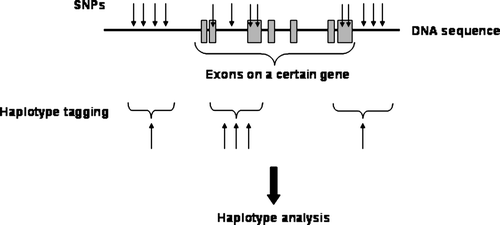
Figure 2. Schematic overview of single nucleotide polymorphisms (SNPs) associated in a certain chromosomal area and the principle of haplotype tagging.
Study designs; candidate genes, pathways analyses, and genome wide association studies
The completion of the HapMap and recent technical advances have made it possible to screen several thousands of individuals for 100 000–1 000 000 genomic variants while comparing affected individuals with healthy control subjects in genome wide association studies Citation[3], Citation[4]. This type of study is agnostic (i.e. hypothesis free) and assumes that genetic variants contribute to disease development in all complex diseases. Recent reviews discuss issues of study design and interpretation of genome wide association studies, GWAS Citation[5]; the current overview includes a discussion of general study designs issues, clinical interpretation of findings, and potential directions of future studies. The majority of GWAS published to date are on common tumour such as breast, prostate, and colorectal cancers Citation[6–9], also reviewed by Easton DF and Eeles Citation[10]. A few studies in cancers with lower frequencies as urinary bladder and thyroid have also been published Citation[11–13]. Alternative approaches to GWAS are candidate gene or pathway based analyses of multiple genes. Candidate genes studies may be considered for a number of reasons including the high costs of GWAS and the lack of statistical power to perform GWAS in rare and lethal cancers, where adequate numbers of samples are difficult to assemble. In addition, for some genes with large variability, the current chip technology does not cover the whole variability in the gene, as the chip only covers 80% of the whole genome genetic variation and are not evenly distributed across the genome. The Human Genome Epidemiology Network (HugeNET) Citation[14] has suggested investigators complete meta-analyses of genetic variants in small studies in a standardised way to be able to compare and identify or discard suggestive loci Citation[15], Citation[16]. This approach has been exemplified successfully in the Interlymph consortium where statistically significant associations between SNPs in TNF, IL10 and risk of lymphoma were identified by pooling data from several existing genetic studies Citation[17]. Huge Net has also published a recent review “Strengthening the Reporting of Genetic Association Studies (STREGA)” Citation[18], with the goal of improving the transparency of reporting and understanding of association studies, so that the evidence of association easily can be compared with other similar studies.
Study design – checklist
Statistical power and sample size
Many of the study design issues that need to be considered when conducting a genetic association study are similar to those that should be considered for any general association study. A checklist of the most important issues is listed in Box 1. Statistical power estimates are closely linked to the sample size, where larger samples size improves power considerably. To analyse single SNPs, a smaller sample size of a few hundred cases could be sufficient, depending on the frequency of the genotype and the size of effect that is of interest. For many of the confirmed identified genetic variants to date the odds ratios are low, ranging from 1.2–1.4, but seldom above that range Citation[19]. This indicates that each genetic variant, even those that are causally associated to cancer risk with high statistical significance, only confers to a small absolute risk effect. To conduct a GWAS, a first discovery set of at least 500 but maybe more optimally 2 000 cases and similar amounts of controls has been estimated as necessary Citation[20]. To address the problem of false positive findings when analysing 600 000 variables, a study design of a first discovery phase and one or more confirmatory stages has been adopted Citation[21]. The size of the discovery phase and the cut-off level for significance to the second and third stage vary in different studies and the best strategy is currently debated and not perfectly delineated yet. Many studies are driven not only by the scientific rationale but also by the high cost of genotyping which forces a lower sample of significant genotypes that are taken forward to the next confirmatory step of the study. Generally SNPs in GWAS with a significance of p < 10 − 7 is considered statistically significant, considering the initial 600 000 genotyped comparisons.
Box 1. Study design issues of association studies for genetic variants
Power estimates
Sample size
Population stratification
Genome wide association or candidate/pathway approach
Hardy Weinberg Equilibrium
Genotyping success and errors
Frequency of studied genetic variants
Functionality of the genetic variants
Replication in independent data sets
Special aspects of rare and lethal cancers
Presently, the most common types of diseases, such as breast, colon and prostate cancer, diabetes and cardiovascular diseases have been studied with GWAS, where it is fairly easy to assemble large data sets of thousands of cases. Many reviews do not address the first obvious obstacle to perform a GWAS; to have adequate sample sizes. For rare disease, genetic association might be stronger and less complex, as shown in the case of chronic lymphocytic leukemia with odds ratios of 1.4–1.5 Citation[19]; for these diseases, smaller sample sizes are required and candidate genes could potentially be identified through alternative approaches such as meta-analyses Citation[17]. Current research efforts for rare diseases may advance more rapidly through the formation of consortia, to increase communication and collaboration between investigators working on rare diseases. Pooling of smaller, published existing studies could be a fruitful way of identifying a network of researchers that are interested in the same diseases. The collaboration of interested investigators is necessary to accomplish the huge effort of collecting rare cancer cases prospectively to allow for the joint analyses of larger sample sets Citation[14]. For very lethal cancers, such as glioblastoma and pancreatic cancer, there is a short time window for the recruitment of patients and the collection of blood samples for genetic association studies.
Studies of rapidly fatal cancer may recruit patients through a clinic based system or require the use of a rapid case ascertainment system for population-based samples to avoid issues of survival bias in patient recruitment. Combined samples from existing prospective cohorts may be a good, alternative source of samples for genetic analyses of rare lethal diseases, which avoids selection bias by sampling.
There may be obvious evidence of genetic susceptibility in rare tumours, as the example of medulloblastoma where siblings of children with a brain tumour are 3.6 times as likely to develop cancer of the nervous system by age 61 as children without a childhood brain tumour-affected sibling Citation[22], Citation[23]. A genetic predisposition to medulloblastoma has been described for several genetic syndromes including Neurofibromatosis types I and II, Li-Fraumeni syndrome, Gorlin's syndrome, Turcot syndrome and Fragile X disease Citation[24]. Even though the evidence that low penetrant genes might be of great importance for medulloblastoma we were unable to find more than one association study published in the literature Citation[25], due to the challenges in assembling adequate genetic samples for association studies. The single, published study used archival, new-born blood spots as their source of DNA, which can be considered as an alternative source of pre-diagnostic, genetic material for studies of childhood diseases with poor prognosis Citation[26]. In summary, sample sizes in rare cancers are a challenging obstacle to perform association studies.
Population stratification
The frequency of genotypes varies considerably in different populations usually by different ethnic origin and different geographical regions Citation[27]. In Scandinavia, differences in allele frequency are most prominent in the western parts of Finland, although some differences in allele differences are also found in different parts of the Nordic countries Citation[28]. When designing case-control studies, the careful selection of cases and controls must be considered if there is an admixture of different ethnic/geographical origin in case compared to controls, usually called population stratification. In case-control studies, differences in allele frequency between cases and controls may lead to confounding of genetic associations, which may over or underestimate associations. In many current GWAS studies, already genotyped controls are used in the discovery set to make the study cost effective, in some cases re-matching controls on age and gender. This has been accepted as a valid study design for genetic studies, but must be used with careful consideration if the population distribution is different in cases compared to the already genotyped controls.
Genotyping errors
To test if genotyping errors exist in the data set, a rigorous check of the control samples in terms of the known frequencies of the genotypes and testing for Hardy-Weinberg Equilibrium (HWE), this assumes that the distribution of genotypes is stable within the generation and that the frequency of the genotypes AA, Aa, and aa, in the proportions p2, 2pq, and q2 respectively. There are different views if all genotypes violating HWE should be omitted from the association study as that also could be a sign of strong association Citation[29].
False positive findings or true association?
A strength of the statistical association is to correct for multiple comparisons and number of methods have been proposed including Bonferroni and permutation testing. The problem with Bonferroni is that it assumes all values to be independent which is not true for genotypes that partly are correlated to each other. Different approaches for validating observed genetic association have therefore been proposed to distinguish true associations from false positives, which are common due to the high number of comparisons that are typically made in genetic studies Citation[30]. First, if the study is a collection of several case control sets, the consistency of an identified association across individual studies strengthens the causal inference Citation[31]. Second, confirmation and replication of the association in one or two independent data sets is suggestive of a true association Citation[5]. Third and maybe most importantly, are to show functional importance of the polymorphisms in disease biology or that the SNP is in LD that is in close correlation with the true functional SNP. Many studies in GWAS published so far have included both exploratory and confirmatory components, but have not provided evidence that explain the functionality of the identified loci. Often the SNP is merely a flag that there is a functional SNP in the region. The identification of the true variant through fine mapping that actually confirms disease risk is a future task that may lead to the biological understanding of the genetic change. Functional studies to explore genotype and phenotype relationships in different experimental systems may lead to a greater understanding of tumour biology and aetiology; the functional consequences of genetic variation are important research questions that needed to be addressed for the translation of genetic association findings into clinically useful knowledge.
Genetic areas associated with several common tumour types
Many different loci have been detected by GWAS but there especially two genomic areas that have been associated with several different cancers, 8q24 and the TERT-CLPTML1 locus.
Chromosome 8q24
Genome wide association studies have recently identified disease associated loci common to several tumour types. One loci of interest is found at chromosome 8q24 which has been associated with several tumour types including prostate, breast, colon and cancer of the urinary bladder. This genomic area does not harbour any known genes except a single pseudogene; the functionality of the associated genotypes have yet not been disentangled. C-MYC, an oncogene located about 30 kb from the loci, does not appear to explain these associations based on functional studies Citation[32]. Interestingly, fine mapping indicates that associations are due to variants indifferent haplotype blocks suggesting disease risk may result from as many as five different functional variants. Resequencing of the area has identified 442 previously unreported polymorphisms, and provided evidence that the area is highly variable Citation[33]. There is consistent confirmation in independent data sets that the association is true, but currently no functional evidence exists.
TERT-CLPTML1 locus
In a large study including 30 000 cases of cancer and 45 000 controls, a genetic variant at chromosome 5p15.33 (rs401681) that previously had been associated with lung cancer Citation[34], was found to be associated with cancers of the lung, urinary bladder, prostate and cervix. In malignant melanoma the same variant conferred to a protective effect Citation[12]. The loci harbour the TERT gene that is important for telomere length maintenance and chromosomal stability, which provides evidence supporting a biological basis for the association. The results from this Icelandic study however, imply that there is more than one functional genotype that explains the risk association Citation[12]. Future directions could include the identification of possible gene-environmental interactions that impact telomere length and genomic instability.
Clinical implications
One goal of genetic association studies is to gain tools for the prediction of disease risk, which will help guide decision making for disease prevention and surveillance strategies. For example, genetic associations may help to identify high-risk subpopulations to successfully focus screening efforts; for common cancers such as prostate or colorectal cancer, this could have large public health and economic benefits. Some researchers have investigated the clinical prediction value of currently identified genomic variants. The receiver operator characteristics (ROC) curve (AUC, area under curve) is often used as an instrument to estimate how much an additional risk factor contributes to the risk prediction. The analysis shows the relationship between the true positive fraction of a test result and the false positive fraction for a diagnostic procedure that can take on multiple values. For breast cancer, an AUC was calculated after adding the seven most strongly associated breast cancer SNPs to a model which included previously recognized breast cancer risk factors such as age at menarche, age at first child, family history of breast cancer, and personal medical history of breast biopsy. The AUC for this model only improved from 0.607 to 0.632, thereby suggesting the need for a larger set of new markers to be able to improve prediction Citation[35]. The accurateness of using AUC has been debated as it is an abstract measurement based on comparison of pairs and the population attributable risk (PAR) has been suggested as a more interesting and useful tool Citation[36]. Similar small effects have been estimated for colon cancer 8. In prostate cancer a set of five SNPs were tested and estimated to explain 45% of prostate cancer cases, with increasing risk with number of risk alleles Citation[37]. However in that study, no increased association was shown with aggressive prostate cancer. This is a clinically important question as there is a substantial problem with over treatment of PSA screening detected subclinical tumours. In a confirmatory study from the US, the association of the five SNPs was also shown, but no improvement of the ROC curve was found for the prediction of prostate cancer risk or mortality, after accounting for known risk factors such as age, PSA and family history of prostate cancer Citation[38]. A subsequent study investigated seven additional identified prostate cancer associated SNPs (12 in total) and found that these genetic variants in combination with age and family history gave a AUC of 0.65, which is similar to predictions found when using PSA Citation[39]. However the authors acknowledge that the study included a large fraction of non-responders and did not include rigorous methods for excluding cases of subclinical prostate cancer. Therefore, additional genetic markers need to be identified to improve risk prediction and translation into clinical use.
Summary
In summary, studies of genetic variants in large consortia and networks of researchers have recently provided new, important insights into the role of genes in cancer aetiology. These genotypes must be transferred to predictive values to be useful in clinical practice. There are also few studies currently designed to address the genetics of rare diseases and greater efforts are need to assemble large case control series of rare cancers. Further, studies designed to study the functional impact of currently identified loci have to be performed, which may include fine mapping and resequencing of identified areas, studies of gene-environmental interactions, functional studies to understand the biological relevance of genetic changes, and prospective studies that incorporate risk alleles in risk prediction models. A variety of approaches will be necessary to further delineate the exact role of genetic markers with different disease phenotypes.
References
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–61
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009; 10: 241–51
- Saxena R, Voight BF, Lyssenko V, Burtt Np, de Bakker PI, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007; 316: 1331–6
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007; 316: 889–94
- Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA 2008; 299: 1335–44
- Garcia-Closas M, Chanock S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin Cancer Res 2008; 14: 8000–9
- Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson DA, et al. Heterogeneity of breast cancer associations with five susceptibility loci by clinical and pathological characteristics. PLoS Genet 2008; 4: e1000054
- Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 2008; 40: 1426–35
- Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008; 40: 316–21
- Easton DF, Eeles RA. Genome-wide association studies in cancer. Hum Mol Genet 2008; 17: R109–R115
- Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet 2008; 40: 1307–12
- Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet 2009; 41: 221–7
- Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet 2009; 41: 460–4
- Ioannidis JP, Bernstein J, Boffetta P, Danesh J, Dolan S, Hartge P, et al. A network of investigator networks in human genome epidemiology. Am J Epidemiol 2005; 162: 302–4
- Ioannidis JP, Gwinn M, Little J, Higgins JP, Bernstein JL, Boffetta P, et al. A road map for efficient and reliable human genome epidemiology. Nat Genet 2006; 38: 3–5
- Seminara D, Khoury MJ, O'Brien TR, Manolio T, Gwinn ML, Little J, et al. The emergence of networks in human genome epidemiology: Challenges and opportunities. Epidemiology 2007; 18: 1–8
- Rothman N, Skibola CF, Wang SS, Morgan G, Lan O, Smith MT, et al. Genetic variation in TNF and IL10 and risk of non-Hodgkin lymphoma: A report from the InterLymph Consortium. Lancet Oncol 2006; 7: 27–38
- Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, Von Elm E, et al. STrengthening the REporting of Genetic Association studies (STREGA)–an extension of the STROBE statement. Eur J Clin Invest 2009; 39: 247–66
- Di Bernardo MC, Crowther-Swanepoel D, Broderick P, Webb E, Sellick G, Wild R, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet 2008; 40: 1204–10
- Gail MH, Pfeiffer RM, Wheeler W, Pee D. Probability that a two-stage genome-wide association study will detect a disease-associated snp and implications for multistage designs. Ann Hum Genet 2008; 72: 812–20
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunder DJ, Thomas G, et al. Replicating genotype-phenotype associations. Nature 2007; 447: 655–60
- Hemminki K, Li X, Vaittinen P, Dong C. Cancers in the first-degree relatives of children with brain tumours. Br J Cancer 2000; 83: 407–11
- Searles Nielsen S, Mueller BA, Preston-Martin S, Holly EA, Little J, Bracci PM, et al. Family cancer history and risk of brain tumors in children: Results of the SEARCH international brain tumor study. Cancer Causes Control 2008; 19: 641–8
- Narod SA, Stiller C, Lenoir GM. An estimate of the heritable fraction of childhood cancer. Br J Cancer 1991; 63: 993–9
- Searles Nielsen S, Mueller BA, De Roos AJ, Viernes HM, Farin FM, Checkoway H. Risk of brain tumors in children and susceptibility to organophosphorus insecticides: The potential role of paraoxonase (PON1). Environ Health Perspect 2005; 113: 909–13
- Searles Nielsen S, Mueller BA, De Roos AJ, Checkoway H. Newborn screening archives as a specimen source for epidemiologic studies: Feasibility and potential for bias. Ann Epidemiol 2008; 18: 58–64
- Tiwari HK, Barnholtz-Sloan J, Wineinger N, Padilla MA, Vaughan LK, Allison DB. Review and evaluation of methods correcting for population stratification with a focus on underlying statistical principles. Hum Hered 2008; 66: 67–86
- Salmela E, Lappalainen T, Fransson I, Andersen PM, Dahlman-Wright K, Fiebig A, et al. Genome-wide analysis of single nucleotide polymorphisms uncovers population structure in Northern Europe. PLoS ONE 2008; 3: e3519
- Wang J, Shete S. A test for genetic association that incorporates information about deviation from Hardy-Weinberg proportions in cases. Am J Hum Genet 2008; 83: 53–63
- Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: An approach for molecular epidemiology studies. J Natl Cancer Inst 2004; 96: 434–42
- Bethke, L, Murray, A, Webb, E, Shoemaker, M, Muir, K, McKinney, , et al. Comprehensive analysis of DNA repair gene variants and risk of meningioma. J Natl Cancer Inst 2008;100:270–6.
- Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J Natl Cancer Inst 2008; 100: 962–6
- Yeager M, Xiao N, Hayes RB, Bouffard O, Desany B, Burdett L, et al. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet 2008; 124: 161–70
- Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 2008; 40: 1407–9
- Gail MH. Discriminatory accuracy from single-nucleotide polymorphisms in models to predict breast cancer risk. J Natl Cancer Inst 2008; 100: 1037–41
- Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J Natl Cancer Inst 2008; 100: 1432–8
- Zheng SL, Sun J, Wiklund F, Smith S, Stattin P, Li G, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008; 358: 910–9
- Salinas CA, Koopmeiners JS, Kwon EM, Fitzgerald L, Lin DW, Ostrander EA, et al. Clinical utility of five genetic variants for predicting prostate cancer risk and mortality. Prostate 2009; 69: 363–72
- Zheng SL, Sun J, Wiklund F, Gao Z, Stattin P, Purcell LD, et al. Genetic variants and family history predict prostate cancer similar to prostate-specific antigen. Clin Cancer Res 2009; 15: 1105–11