
Abstract
Background
Colombia has a mestizo population and the prevalence of haemoglobin variants varies according to each region, but heterozygous carriers can be found in all of them.
Aim
To characterise sickle cell disease (SCD) haematologically, biochemically, and molecularly, and detect classic haplotypes by DNA sequencing in a group of samples from Bolívar, Colombia.
Subjects and methods
Blood samples were collected after informed consent from volunteers from eight communities in the Bolívar department, plus samples from the Pacific region, Providencia Island, and Bogotá were included. Data were obtained from: (1) haematological analyses; (2) biochemical tests: dHPLC was used to determine haemoglobin (Hb); and (3) DNA sequencing data through five SNPs.
Results
101 samples were identified by rs334 through Sanger’s Sequencing, structural haemoglobinopathies HbAS (34.65%), HbSS (2.97%) and HbAC (1.98%) were found. When contrasting the Hb identification results between SNP rs334 Vs. dHPLC/Isoelectric Focusing (IEF), a coincidence was found in 39/43 samples analysed, therefore, when comparing these techniques, a significant correlation was found (Pearson’s correlation coefficient r = 0.998). 26 samples previously analysed by rs334 were classified into classical haplotypes CAR (50.0%), BEN (30.76%), CAM (7.69%), SEN (3.84%), and ATP-I (7.69%).
Conclusions
SCD characterisation and SNPs-based classification through Sanger’s DNA sequencing have not been performed before in Colombia. The results of this work will make it possible to expand the data or records of carriers and those affected, which will benefit patients and their families.
Introduction
Sickle cell disease (SCD) is a haemoglobinopathy due to a substitution of a single nucleotide, a change from adenine to thymine in the β chain gene for the synthesis of haemoglobin (HBB: c.20 A > T, rs334), on chromosome 11, causing a substitution of the amino acid glutamic acid for valine in the sixth position of the protein (Rosero and Bermúdez Citation2012; OMIM Database Citation2022). In the homozygous state the disease manifests itself, at any age, acutely with severe anaemia, loss of red cell deformability, susceptibility to serious bacterial infections, and ischaemic vaso-occlusive accidents (Schaefer and Thompson Citation2016). The disease can appear at any age (Orphanet Citation2021). The World Health Organisation (WHO) has estimated that around 7% of the world population carries a genetic defect in haemoglobins and that each year around 300–400,000 severely affected children are born between homozygotes and heterozygotes; of these about 50% involve haemoglobin S (HbS) (Ibarra-Cortés et al. Citation2019).
Patients with SCD show a wide spectrum of clinical manifestations from mild anaemia to severe forms of the disease, including lethality (Fatima and Mussaed Citation2015; Aghajani et al. Citation2016). Due to the heterogeneity of clinical manifestations detected between patients with similar genotype, whether homozygous or heterozygous (asymptomatic), SCD is currently identified as a multifactorial disease (Ibarra-Cortés et al. Citation2019).
According to the National Organisation for Rare Disorders in the United States (NORD), the frequency of SCD varies from country to country. Mutations in the HBB gene (β-globin cluster) are common in people from Africa, the Mediterranean, the Middle East, India, and in people from the Caribbean and regions of Central and South America, but can be found in individuals of any ethnic origin (Bender Citation2019).
Colombia has a mestizo population (Díaz-Matallana and Martínez-Cruzado Citation2010; Yunis and Yunis Citation2013; Noguera et al. Citation2014), and the prevalence of haemoglobin variants varies according to each region, but heterozygous carriers can be found in all of them (Bernal et al. Citation1995; Romero-Sánchez et al. Citation2015). In the beta globin gene HBB (MIM: 141,900), the sickle (MIM: 603,903) allele βS is under balancing selection because of heterozygote advantage, which results from protection against malaria caused by Plasmodium falciparum and recessive lethality (Shriner and Rotimi Citation2018).
The β-globin gene cluster is located along a 60,000 bp segment of the short arm of chromosome 11 (11p15.5) and contains five functional genes (β, δ, Gγ, Aγ, and ε) and one pseudogene. The genes are arranged in the order in which they are expressed during different developmental stages (Tripathi Citation2022). The characterisation of classic haplotypes associated with the β-globin cluster has allowed us to elucidate cultural and ethnic mosaics of certain populations and to clarify, at least partially, the reason for the clinical heterogeneity of SCD, due to the fact that depending on the haplotype, clinical pictures of severe or moderate SCD may occur (El-Hazmi et al. Citation1999; Durán et al. Citation2012; Shaikho et al. Citation2017; Shriner and Rotimi Citation2018; Adekile et al. Citation2021).
The general objective of this work was to characterise SCD haematologically, biochemically and molecularly and detect classic haplotypes by means of DNA sequencing in a group of Colombian individuals in order to: determine the presence of patients with SCD and sickle cell trait (SCT) in the individuals analysed; describe the haematological characteristics of the samples detected with SCD or SCT; identify SCD through specific molecular markers and DNA sequencing in the collected samples; and determine the frequency of the haplotypes associated with the β-haemoglobin gene (βS gene samples) in the individuals analysed.
Materials and methods
Ethics
The Research Ethics Committee of U. Sabana in session No.75 (May 22nd, 2019) approved this research project, stating that it meets the quality requirements in research and complies with the ethical criteria and the current provisions that regulate the subject in Colombia. Written informed consents were applied to the voluntary individuals, from whom the blood samples were collected.
Sampling type
Convenience (non-probabilistic); in this sampling method, the units are selected because they are convenient for the researcher (Ruiz-Morales and Gómez-Restrepo Citation2015). The convenience is geographical, logistical, and due to the availability of samples, that is, depending on the participants who agree to participate in this study.
Study population
Blood samples were collected from unrelated individuals in different presentations from eight (8) communities from Bolívar department-Colombia, as follows: (a) Local Hospital at Calamar Jun-Jul 2022 (n = 25); (b) Mompox Health Centre Nov. 2022 (n = 23); (c) Cartagena de Indias (2022-II) at Universidad del Sinú (n = 130); (d) Pre-existing samples Bolívar (April 2010) at Universidad del Sinú, Cartagena Campus, and revised in April 2022 (n = 130), which are described below as follows: Municipalities of the “Canal del Dique” [n = 14: Arroyohondo (2), Calamar (3), Maria La Baja (1), Turbana (3), Turbaco (3), Cartagena (2)]; additionally samples from Palenque de San Basilio (n = 17); Calamar (n = 78); samples from the “Pacific” and Providencia Island (n = 18) [9 Chocó Nuquí, 2 Bahía Solano, 2 Bocas Guangüí-Cauca, 5 Providencia (Prov.)]; and (e) filter paper samples provided by Pregen in April 2022 (n = 4), from newborns at Clinics-Bogotá, with abnormal Hb. Plus, two (2) samples of Lab. Controls from Bogotá were included. The age and sex information of these samples are detailed in .
Table 1. Age and sex data of the population groups analysed.
Basic haematological tests
Haemoglobin, mean corpuscular volume, and mean corpuscular haemoglobin concentration (fresh cells). IV Generation tests were carried out at Clinical Laboratories from Bolívar. Likewise, the parameters of haematocrit, leukocytes, platelets, and reticulocytes were considered ().
Table 2. Haematological results Calamar (n = 24), Haemoglobin S (n = 25).
Table 3. Haematological results Mompox (n = 23).
Table 4. Haematological results control genetics labTable Footnoteb.
Biochemical tests
Denaturing High Performance Liquid Chromatography (dHPLC) was used. Haemoglobin (Hb) chromatograms were processed at the Pregen Genetic Medicine Laboratory (Bogotá). Ultra 2-215, Trinity GeneSys Assay V.1.2.2, Instrument Serial No. 100682 was used. The Glucose 6 Phosphate Dehydrogenase (G6PD) deficiency test was added to the analyses.
Genomic DNA isolation
DNA extractions were carried out in the Genetics Lab of U. Sabana. Different protocols were used, as indicated below: (A) for whole blood samples, the protocol of salting out method was used, for centrifuge tubes (15 ml) and for microtubes (1.5–2 ml), which had been adapted from Sambrook and Russell (Citation2001), and was modified during the Post-Doc of M.D.M. [Supplements S1.1–S1.3]; (B) for blood samples on filter paper, a fusion of extraction protocols composed of Chelex-100 resin (10%) and salting out was used: (a) GE Healthcare (2010); (b) Díaz-Matallana and Martínez-Cruzado (Citation2010); and (c) Díaz-Matallana (Citation2015), Díaz-Matallana et al. (Citation2016) (Supplements S2 and S1.3).
Genomic DNA was isolated from 179 blood samples (161 Bolívar, 13 Pacific and 5 Providencia Island), defined as follows: 24 Calamar (fresh); 14 “Canal del Dique” (microtubes); 17 Palenque de San Basilio [SBP, microtubes]; 78 Calamar (microtubes); 23 Mompox (fresh); 4 Pregen (filter paper), CAL 11 (filter paper); 13 Pacific and 5 Providencia (n = 18). The isolated DNAs were verified by agarose gel electrophoresis stained with SYBR Safe (Invitrogen®) and then visualised in the GelDocXR (Bio-Rad®). DNAs were quantified by NanoDrop 2000c (Thermo Scientific®).
DNA amplification
Initially, the amplification of the SNP rs334 was verified, according to Fatima and Mussaed (Citation2015), and the published SNPs data reviewed (Shaikho et al. Citation2017, Shriner and Rotimi Citation2018, dbSNP-NCBI Citation2022). Subsequently, several primer pairs were designed with Primer-Blast Software (dbSNP-NCBI Citation2022) and OligoAnalyzer Tool (IDT, USA). The selected and implemented primers (5 pairs) for PCR and sequencing, which generate a product >200 bp, are displayed in . The design of the primers, standardisation of the PCR conditions for all the primers and the respective amplifications, were carried out at U. Sabana by the post-doc M.D.M. PCR reactions were set up in a MAXYGENE thermocycler. Each PCR reaction consisted of the following molecular biology reagents: GoTaq® Flexi Buffer Promega [1X], MgCl2 [2.5 mM], dNTPs Promega [2.5 mM], Forward & Reverse Primers [1 µM], GoTaq® DNA polymerase Promega [2 U], DNA template [< 200 ng/25 µl], sterile deionised distilled H2O, to 25 µl final volume. The PCR products were checked from agarose gel electrophoresis, a 100 bp Hyperladder (Bioline®) was included and visualised by GelDocXR (Bio-Rad®).
Table 5. Description of oligonucleotides designed and selected for amplification (PCR) and Sanger’s SequencingTable Footnotea.
DNA sequencing
Sanger sequencing and purification was performed by Macrogen Inc. Seoul, South Korea (Supplements S3(a,b)) mediated by CorpoGen (Bogotá, D.C). The “ABI PRISM® BigDyeTM Terminator Cycle Sequencing” kit, capillary electrophoresis, and an ABI PRISM® 3730XL genetic analyser (96 capillaries) were used.
Validity and reliability criteria
Only good quality DNA sequences were considered, to ensure the reliability of the data, which influences the SNPs identification and haplotype assignment (Supplements S3(a,b)). Regarding validity, the repeatability (2–4 reps each) and reproducibility of the amplified products and of the DNA sequences were evaluated. In each PCR reaction a negative control was included to monitor possible contamination.
Data analysis
For various haematological data and age by population group, the mean was calculated using the MaxStat Lite v.3.60 program (©2015). The Hb result (dHPLC) was analysed by simple frequency calculation. The mean of the G6PD deficiency test for Calamar and Cartagena was calculated with this program. Multiple alignment of DNA sequences and electropherograms were performed using the programs Sequencher® v.5.0 (©1991–2011) and BioEdit v.7.2.5 (©1997–2013). Sequence data allowed the frequency of Hb type to be determined using SNP rs334.
The sequence data (rs334) were contrasted with the chromatogram results to determine the concordance (%) of the results in Hb identification by simple counting. Additionally, Pearson’s correlation test was applied to structural haemoglobinopathies of 43 comparable samples, between SNP Vs dHPLC/IEF methods, as well as quantitative Hb through MaxStat Lite v.3.60 (©2015). The sequence data of the other four SNPs facilitated the determination of the classical haplotypes, as well as their respective frequencies. With the Network v.10.2.0.0 program (©2004–2023) a phylogenetic network was built with the data obtained from haplotypes.
Results and discussion
shows the age and sex data of the populations studied, from 190 blood samples from the department of Bolívar, Colombia and six (6) samples from Bogotá, D.C.
Haematological results
and show the mean of haematological parameters for the Calamar and Mompox population groups. Haematological parameters include haemoglobin concentration (HBG), haematocrit (HCT), mean corpuscular volume (MCV), mean haemoglobin concentration (MCHC), leukocytes (WBC), platelets (PLT) and reticulocytes. In the results of haemograms, variation is observed in the reference values considered by clinical laboratories for the processing of Calamar and Mompox samples.
In the mean of reticulocyte data obtained in Calamar (), a higher value is observed with respect to the mean of data of this parameter in Mompox () (Tukey test: p > .05 Non-Significant (N.S.) difference). On the other hand, it is evident that the mean leukocyte data is below the normal reference value (Tukey test: p > .05 N.S. difference).
It should be noted that the results of the haemogram for the CAL18 (Calamar) sample showed severely decreased values of HGB (8.5), erythrocytes (2.81), and HCT (24%), while they indicated increased values of leukocytes (12.19) and platelets (557), as well as the highest value of reticulocytes (10.8%) of the Calamar group (). This pattern in haematological parameters can be interpreted as the presence of SCD (HbSS). Likewise, an elevated reticulocyte count is present in patients with congenital or acquired haemolytic anaemia, in normal newborns, and in chronic blood loss (Gómez-Gutiérrez and Casas-Gómez Citation2014).
The haematological parameters of Calamar indicated that all subjects heterozygous for Haemoglobin S (CAL9, CAL14, CAL17) have normal values of Hb, mean corpuscular volume (MCV), and mean corpuscular Hb concentration (MCHC), regardless of age and sex. Likewise, the reticulocyte values (%) were within the normal range established by the Clinical Lab ().
shows the results of the blood count for a laboratory control, where the results are within normal ranges, however the values of HGB (13.5) and HCT (39.5%) are higher than those obtained in Calamar and Mompox groups (Tukey test: p > .05 N.S. difference). Reticulocyte data were not considered.
Biochemical results – haemoglobin chromatograms
shows the frequencies of the results of the Hb types for the Calamar group, according to the results of the chromatograms (dHPLC), where 22 individuals were identified with HbA (normal) and 3 individuals were identified with HbAS, known as a heterozygous trait (CAL11, CAL14 and CAL17).
Figure 1. Multiple Compared Graph of Haemoglobin (Hb) frequencies obtained for communities of Bolívar, Colombia. (A) Frequencies of Hb Types obtained for Calamar group from Hb Chromatograms; (B) Frequencies of Hb Types obtained for Cartagena (2022) from Hb Chromatograms in this work; (C) Frequencies of Hb Types obtained for Calamar group with data verified at the sequencing level (SNP rs334) in the present study; (D) Frequencies of Hb Types obtained for the group of municipalities of the “Canal del Dique” from sequencing data (SNP rs334) in the present research; (E) Frequencies of Hb Types obtained for Palenque de San Basilio (SBP) from sequencing data (SNP rs334) in the present work. The population showed in is the same (Calamar), but with different analyses.
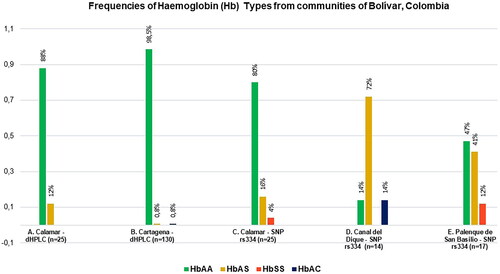
However, two individuals showed confusing results on Hb chromatograms, CAL9 and CAL18, as both were identified with HbA, but the data were showing another possible Hb assignment as follows, CAL9: 39.5% A, 35.1% S; and CAL18: 62.5% S, 16.0% A. Therefore, these results were corroborated at the molecular level with the SNP rs334, in order to clarify the type of Hb for such individuals (Supplementary Table S4).
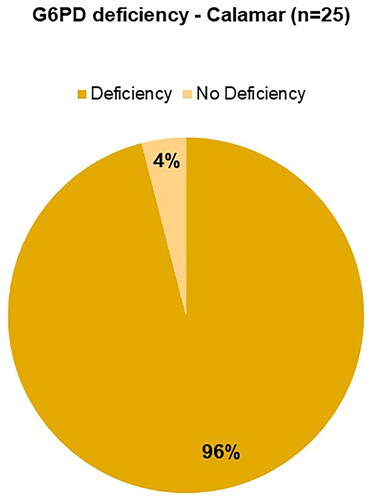
shows the results of the G6PD deficiency test for the Calamar group, where 24 individuals present the deficiency (abnormal) and only 1 individual (child: CAL5) does not have a deficiency (2.7 U/gHb). The mean of these data was 1.85 ± 0.62 (SD) (Ref. Normal Adult: 5.5–20.5 U/gHb, Ref. Child: >2.6 U/gHb). G6PD is an enzyme involved in glucose metabolism. Its deficiency causes Hb precipitation and changes in the erythrocyte cell membrane, which can lead to intense haemolysis. Decreased values can be found in haemolytic anaemia (Medline Plus Citation2023).
Figure 2. Frequencies of the Glucose 6 Phosphate Dehydrogenase (G6PD) test for Calamar, Bolívar in this study.
shows the results of the Hb types of the samples from Cartagena de Indias, in which 128 individuals were classified as HbA, one (1) individual as HbAS (444698) and one (1) individual as HbC (450401). On the other hand, the result of the G6PD deficiency test for the Cartagena group (2022, n = 130), indicated that 100% present the deficiency. The mean of these data was 1.23 ± 0.51 (Normal Ref. value: 5.5–20.5 U/gHb), which is much lower than that of the Calamar group.
Alvear et al. (Citation2012), estimated the incidence of haemoglobinopathies in Cartagena. The methodological design proposed a descriptive study on 1729 cord samples processed by IEF, 4.5% were heterozygous for haemoglobin S, 1% were heterozygous for haemoglobin C, and 0.1% were double heterozygous for S-C (Romero-Sánchez et al. Citation2015). When contrasted with the results of the Hb chromatograms illustrated in of this work (Cartagena n = 130), the less frequent values of HbAS and HbAC are similar.
Molecular markers and DNA sequencing
rs334 SNP identification
SNP rs334 was used as a molecular marker to identify SCD (Fatima and Mussaed Citation2015; Shaikho et al. Citation2017; Shriner and Rotimi Citation2018; dbSNP-NCBI Citation2022) (). and Supplementary Table S4 show the results with this marker for 101 samples from Colombia.
Table 6. Frequency of structural Haemoglobinopathies found in samples from Colombia (n = 101), based on the SNP rs334 through Sanger’s SequencingTable Footnotea.
The Hb types were verified for the Calamar group (n = 25), which are shown in , , and Table S4. In these results, the following types of Hb were found: 20 HbA individuals, 4 HbAS individuals (CAL9, CAL11, CAL14 and CAL17) and 1 HbSS individual (CAL18), therefore, the frequencies of the Hb types obtained by SNP rs334 changed in contrast to dHPLC analysis in , with data clarified and better defined by SNP rs334 in .
It should be noted that the reference by Fatima and Mussaed (Citation2015) was initially taken into account for the amplification and sequencing of the SNP rs334. However, with the results obtained, very short sequence fragments (133 bp) were observed, therefore, the quality of the sequences was not optimal. For this reason, new primers were designed to amplify and sequence this SNP (), which produced a 232 bp fragment and allowed the generation of PCR products and good quality sequences in various samples in this work ( and , Table S4). and Table S4 show the results of the identification of SNP rs334 (np. 5,227,002 -Chromosome 11) by DNA sequencing, that is, a total of 101 samples (Pregen, Calamar, “Canal del Dique” Municipalities, Palenque de San Basilio, Mompox, Pacific, Providencia Island and two Lab controls) identified with SNP rs334, for a total of 262 DNA sequences analysed containing this SNP [the samples from Cartagena 2022 (n = 130) and Calamar 2010 (n = 78) were not included in this analysis]. The reference sequence is in the range 5,226,901–5,227,081 (Accession NC_000011.10 – NCBI) with a size of 181 bp for this SNP.
Table 7. Results of classical haplotypes associated with the β-globin cluster.
The SNP rs334 sequence results are contrasted with the Hb chromatogram results for the samples from Pregen (n = 4) (Table S4), Calamar (n = 25) (), and Canal del Dique Municipalities (n = 14) ().
Regarding the samples from Palenque de San Basilio, with the data of the SNP rs334, the following haemoglobin types were found: 8 individuals with HbA, 7 individuals with HbAS (heterozygous) and 2 individuals with HbSS (homozygous) (, Table S4, and ). In Supplements S3(b), an example of sequence alignments and electropherograms with SNP rs334 for an individual detected as homozygous (SBP-272) is illustrated.
Regarding Mompox, the 23 samples turned out to be HbA, that is, normal ( and Table S4). The Pacific results corresponded to six (6) HbAS and six (6) HbA. While in the samples from Providencia Island, five (5) HbAS were obtained (Table S4).
When contrasting the results between SNP rs334 vs. Hb dHPLC/IEF, a coincidence was found in the results in 39/43 samples analysed (excluding those from Palenque de San Basilio, Mompox, Pacific, Providencia Island, and the two Lab control samples), therefore, a concordance of 90.7% using simple counting. Likewise, in the sequences of the analysed fragments, an additional polymorphism was detected in np. 5,227,013, which when reviewed in dbSNP (dbSNP-NCBI Citation2022), corresponds to the SNP rs34058656 with clinical significance of β-thalassaemia and was detected in several samples (Table S4). For example, individual 155199 (Pregen) was confirmed to have β- thalassaemia with the SNP rs34058656 (Table S4).
When calculating Pearson’s correlation test for the structural haemoglobinopathies of 43 comparable samples between the SNP Vs. dHPLC/IEF methods, a correlation coefficient r = 0.998 (standard error 0.040) was found, where a significant correlation was evidenced (p value = .0016, 95% significance level). When calculating Pearson’s correlation test for the quantitative haemoglobins of 43 comparable samples between the SNP Vs Chromatograms/IEF methods, a correlation coefficient r = 1.0 was found. A Pearson’s correlation coefficient between values of 0.80–1 indicates a very strong correlation (MaxStat Citation2015), in this case between the mentioned techniques. Note that in , frequencies of Hb types based only on the SNP rs334 were presented.
On the other hand, in the identification of haemoglobin C (HbAC), there is a particular change in np.5,227,003 (T, C), associated with the SNP rs33930165 (SNPedia Citation2022–2023; OpenSNP Citation2023). HbC was found in individuals from municipalities of the “Canal del Dique”, Bolívar (85: Arroyohondo, 378: Turbaco) (, Table S4, and ). These molecular data are consistent with Pregen’s previous IEF identification of Hb. In the same way, as mentioned in , in recent sampling from Cartagena an HbAC individual was also found, although identified only by dHPLC.
Taking into account the work of Bernal et al. (Citation2010), based in Buenaventura, these authors identified 23 FAC (foetal AC) newborns in 399 samples collected. Haemoglobin C is more frequent in Western Africa, and it is associated with minor symptoms related to malaria infection (Rihet et al. Citation2004). Homozygous C has lower complications related to SCD when compared to homozygous S (Varela et al. Citation2013). They may even be asymptomatic into adulthood, although the retinopathy related to SCD is more frequent with haemoglobin C (Abdalla Elsayed et al. Citation2019).
Regarding the results of , in this work three (3) homozygous individuals (HbSS) were identified from Calamar (1) and Palenque de San Basilio (2), Bolívar ( and Table S4], that is, they belong to the Colombian Atlantic Coast. When contrasting with other studies, such as for e.g. Alvear et al. (Citation2012), no homozygote was detected in Cartagena, while Rosero and Bermúdez (Citation2012), identified one (1) HbSS individual in the Guajira -Atlantic Region- and three HbSS in the Chocó-Pacific Region.
Regarding heterozygous individuals (HbAS), in this work 35 individuals from seven (7) communities of Bolívar were identified, as well as from the Pacific, Providencia Island and Bogotá (, Table S4, and ). This contrasts with the results of Rosero and Bermúdez (Citation2012), who detected 151 HbAS individuals from the Atlantic Coast, Pacific Coast, San Andrés Island, and Putumayo.
In the case of Bernal et al. (Citation1995), for the case of San Andrés and Providencia, in a sample of 544 individuals, both HbAS (6.8%) and HbSS (0.2%) were found. While in our work five (5) HbAS individuals were detected in Providencia Island ( and Table S4).
In Table S4 of this study, one can also visualise the presence of quantitative haemoglobins, such as A-β thalassaemia heterozygotes (26/61 HbA) and S-β thalassaemia heterozygotes (11/35 HbAS). This type of haemoglobin was detected in this work in a proportion equivalent to that reported by Romero-Sánchez et al. (Citation2015); A-β thalassaemia heterozygotes (13.91%) and S-β thalassaemia heterozygotes (6.07%) in 1407 individuals. According to Thein and Menzel (Citation2009), SCD and β-thalassaemia, caused by lesions affecting the HBB (β globin gene), are the most common human genetic disorders worldwide, and represent a major public health problem.
As both foetal (HbF, α2γ2) and adult Hb (HbA, α2β2) contain a globin chains, the switch from foetal to adult Hb production is essentially that of the replacement of HBG (γ globin) with HBB (β globin) gene expression. By six (6) months after birth, HbA becomes the major Hb and mutations which affect HBB, e.g. β-thalassaemia and SCD, become clinically apparent. HbF, which comprises <5% of the total Hb at 6 months, continues to fall, reaching the adult level of <1% (Jiménez-Cobo et al. Citation2021).
SCD is a public health problem due to the high morbidity and mortality rate on the African continent, in the Mediterranean Sea region, in the Middle East, in India, and in every population where there is migration of individuals of African descent. The high prevalence of this disease, together with the presence of thalassaemias and G6PD deficiency, is due to a natural selection process exerted by malaria on individuals carrying these genetic disorders (Ibarra-Cortés et al. Citation2019). In this sense, it is related to this work according to the frequencies of structural haemoglobinopathies detected, heterozygous and homozygous ( and Table S4), the presence of thalassaemias ( and Table S4) and a marked deficiency of G6PD, especially observed for Calamar (96% G6PD deficiency) () and Cartagena (100% G6PD deficiency) [not shown].
On the other hand, it is not only the distribution of malaria that determines the prevalence of haemoglobinopathies in a geographical area, there are also other factors to consider such as ethnicity, consanguinity, and geographic isolation (Jiménez-Cobo et al. Citation2021).
It is well known that G6PD deficiency is an X linked trait, at the cytogenetic location Xq28 (MIM # 305900). The most common types are Mediterranean, which affect those of Jewish descent, and type A, to the Afro-descendant population. In the United States the frequency is 11% in the African American population, and there is a potential number of women within this population of 20%. It is also found in people of the same population in India, Japan, Southeast Asia, and some parts of the Mediterranean (Gómez-Gutiérrez and Casas-Gómez Citation2014).
It should be noted the power of definition provided by Sanger’s DNA sequencing is considered a robust testing strategy able to determine whether a point mutation or small deletion/duplication is present (CDC Citation2015; Gomez and Kurf Citation2018). In this sense, it deals with SNPs and changes in a nucleotide position in a sequence fragment, while Hb IEF/dHPLC are separation methods of Hb types at a protein level. However, dHPLC offers a superior resolution and more rapid results than the IEF method (Joutovsky et al. Citation2004). Regarding the fragment generated by the SNP rs334 with a size of 232 bp, it allowed the detection not only of the SNP of main interest located at np. 5,227,002 (SCD), but also of other nucleotide positions associated with reported SNPs such as np. 5,227,013 (rs34058656: B-TAL), and np. 5,227,003 (rs33930165: HbC).
However, the four (4) samples supplied by Pregen (Bogotá), previously identified as abnormal haemoglobins on filter paper by dHPLC, played a very important role for the Hb determination via sequencing-SNPs and as a standard of comparison between techniques ( and Table S4). Likewise, previous information on the Hb types was obtained from the samples from Calamar, Cartagena (dHPLC) and Municipalities of “Canal del Dique” (IEF) by Pregen.
These results show the importance of DNA sequencing and SNPs identification to clarify confusing data, complement data and optimise the quality of diagnoses of SCD and thalassaemias (, , and Table S4), among other haemoglobinopathies, either in an early development of the disease, or, for the determination of heterozygous/carriers (Díaz-Matallana et al. Citation2021).
Classic haplotypes spanning the β-globin gene cluster
For the determination of the classic haplotypes, we worked with four (4) SNPs taking into account what was reported by Shaikho et al. (Citation2017), Shriner and Rotimi (Citation2018), and Adekile et al. (Citation2021), for a total of 311 DNA sequences analysed. The SNPs markers were designed in Primer-Blast – NCBI (see Methods). The chromosomal background of the βS allele has been classified in five haplotypes, Arabian/Indian, Benin, Cameroon, CAR, and Senegal, named after ethno-linguistic groups or geographic regions, as well as a sixth category for “atypical” haplotypes (Hanchard et al. Citation2007). Such haplotypes are named after the geographical areas where they are more prevalent. Haplotype analysis of the βS allele helps us to trace the origin of this Hb variant in a region (Aghajani et al. Citation2016).
shows complete haplotype results for 26 individuals: 7 from Calamar, 3 from Pregen, 5 from Palenque de San Basilio, 9 from “Canal del Dique” and 2 Lab controls, where 5 haplotypes were detected: CAR or Bantu (50%), BEN (30.76%), CAM (7.69%), SEN (3.84%) and Atypical-I (7.69%, Lab control) (). The AI or Arabian-Indian haplotype was not detected in the samples analysed.
Figure 3. Classical Haplotype frequencies for individuals of Calamar, Bogotá-Pregen, Palenque de San Basilio, “Canal del Dique” and two controls, from four (4) SNPs (), in this work.
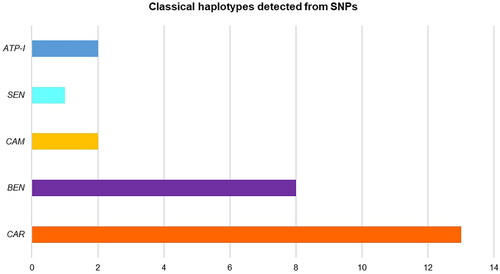
In this study, three HbSS individuals: CAL18, SBP-272, and SBP-313 were classified with the Benin (BEN) haplotype, as well as two HbAS individuals: Pregen 4 (156436) and Turbana (308) were identified with the BEN haplotype and one (1) individual HbC: Arroyohondo (85) was determined as BEN (). Of the 38 individuals analysed, 12 could not be classified into the haplotype, particularly due to SNP rs968857, which presented greater difficulty in amplification and obtaining sequences, compared to the other SNPs. Eleven of these samples presented more difficulty in amplifying and/or generating the sequence since the sampling was ≥ 10 years old, and also perhaps the area where the SNP rs968857 is located (5′ δ HBD) is a more delicate or sensitive region than the regions of the other SNPs.
Studies suggest that the largest proportion of individuals arriving in the Americas from Africa originated from Benin – BEN (70%) and small proportions from the Central African Republic – CAR (17%) and Senegal – SEN (13%) (Muñiz et al. Citation2000; Durán et al. Citation2012).
In the individual CAL 18 (Calamar, Bolívar), a great relationship was evidenced between the haematological results: very high blood count parameters and reticulocytes 10.8%; homozygous Hb (HbSS); was classified as BEN haplotype; G6PD deficiency (2.4 U/gHb) (); and in addition, CAL 18 presented jaundice at the phenotypical level.
In relation to the other two individuals SBP-272 and SBP-313 (Palenque de San Basilio) identified with HbSS and BEN haplotype, the haemogram and reticulocyte tests had not been performed on this population group, since they were pre-existing samples in U. Sinú; thus, in this work only SNPs data were obtained for these individuals.
The other individuals identified as BEN haplotype correspond to heterozygous: Pregen 4 (FAS), Turbana-308 (HbAS), and Arroyohondo-85 (HbAC), for whom Hb Chromatogram data were available, and molecular data were obtained by SNPs in this study ( and Table S4).
It is noteworthy that CAL4 and CAL12 individuals, who were classified as BEN haplotype, corresponded to the HbA or normal type, but when related to the haematological data, slightly decreased values of HGB (10.6), HCT (33.3%) were found, VCM (71.1), MCHC (31.8) and slightly high reticulocytes (2.2%) for CAL 4; as well as higher reticulocytes (2.6%) for CAL 12 (). These individuals presented G6PD deficiency (CAL4: 1.5 and CAL 12: 1.7 U/gHb) ().
On the other hand, the Calamar individuals identified as heterozygous (HbAS), which were classified with the following haplotypes: CAL 9 (CAR), CAL 17 (CAR) and CAL 14 (SEN) (), were related to haematological results, which were within normal values, but with deficient or diminished G6PD evidence ().
The presence of a particular haplotype has been correlated with the severity of symptoms in individuals with SCD and is thought to also alter the clinical course of SCT (Lyra et al. Citation2005). The clinical presentation of the disease is more severe in those homozygous for HbS who inherit Benin (BEN) and Bantu (CAR) than in those who inherit Senegal and Bantu; Cameroon or CAM is rare and has intermediate severity (Durán et al. Citation2012).
According to El-Hazmi et al. (Citation1999), in the study group of SCD patients (n = 126), the BEN haplotype was found in patients of severe disease based on clinical and haematological parameters. Most Syrians and Jordanians had the BEN haplotype and severe disease, also all Saudi patients where the disease is severe had the BEN haplotype. In line with Powars et al. (Citation1990), patients with the BEN and CAM haplotypes had severe and mild phenotypes, respectively. Interestingly, regarding SCD, many modifiers and cofactors have been described (Lettre Citation2012; Ibarra-Cortés et al. Citation2019).
In Colombia, another study carried out in a coastal population with SCD found the Bantu or CAR, BEN, and SEN haplotypes, like the frequencies found in Brazil (Cuéllar-Ambrosi et al. Citation2000). In the present study, the highest frequency of the CAR haplotype (50%) stands out, as in a previous study where the frequency of the CAR haplotype was the highest (58% n = 29 children, 55.5% n = 22 adults) (Cuéllar-Ambrosi et al. Citation2000; Durán et al. Citation2012).
In Medellín, Colombia, patients with SCD were studied. The analysed chromosomes (n = 92) revealed typical African haplotypes, 55.5% CAR, 34.8% BEN, 4.3% SEN and 5.4% CAM. These data agree with reports indicating that most of the African slaves brought to Colombia come from Angola (Bantu population) and the island of Sao Tomé in the Gulf of Benin (Central-West Africa) (Cuéllar-Ambrosi et al. Citation2000; Romero-Sánchez et al. Citation2015).
contrasts the frequencies of classic haplotypes from several studies, including the present work (), where the similarity of haplotype frequencies from the studies from Panama, Colombia and Brazil can be observed, especially in the proportion of CAR, followed by BEN. It should be noted that this work is very similar to the work of Cuéllar-Ambrosi et al. (Citation2000).
Table 8. Comparison of classical haplotypes found in eight (8) Studies from the Americas including our study.
On the other hand, in Venezuela and Cuba the order of haplotypes found was Benin, Bantu and Senegal (Muniz et al. Citation1995; Moreno et al. Citation2002) (). In Mexico, the presence of Benin and Bantu haplotypes has been reported (Old Citation2003). According to another study on patients with SCD in the U.S.A. (Cruz et al. Citation2019), the order of frequencies of classic haplotypes found were BEN (63.3%), Atypical (11.7%), CAR (10.0%), CAM (8.3%), SEN (6.7%), and high levels of divergence are reported with respect to populations from Brazil ().
This work is the first in Colombia that incorporates the use of SNP markers through Sanger’s DNA sequencing for the detection of SCD, as well as for the classification of the classic haplotypes associated with the β-globin cluster (, Table S4; and Figure S3(a,b)), since the previously reported works on SCD were based on PCR-RFLPs markers (Bernal et al. Citation1995; Cuéllar-Ambrosi et al. Citation2000; Bernal et al. Citation2010; Alvear et al. Citation2012; Durán et al. Citation2012; Rosero and Bermúdez Citation2012; Fong et al. Citation2013; Ortega et al. Citation2015; Romero-Sánchez et al. Citation2015), or PCR-RFLPs and TaqMan procedure (Fong et al. Citation2015), likewise most of these publications were previously reviewed by Díaz-Matallana et al. (Citation2021).
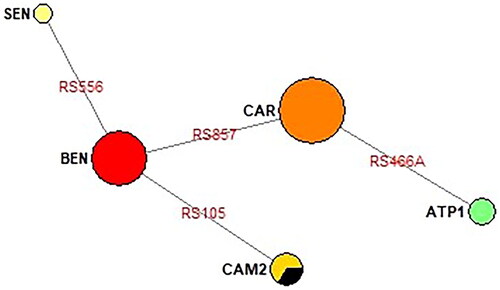
Figure 4. Phylogenetic network of classical haplotypes identified in this study built using Network v.10.2.0.0 (Median joining network). Representation of the haplotype’s relationship based on sequence data from . The SNPs names are shown in red, abbreviated version (last three numbers, and ). The central node is CAR haplotype, and the secondary node is BEN haplotype, from the latter SEN and CAM are derived. The Reference sequence is included in CAM haplotype (black color). Samples from Pregen, which correspond to newborns at Clinics in Bogotá, seem to come from the Atlantic Coast, since they fit quite well into the phylogenetic network: 2 CAR and 1 BEN (). ATP1 is an atypical haplotype ().
Our results evidenced the importance of SNPs identification by Sanger’s DNA sequencing to clarify confusing data, complement data and optimise the quality of diagnoses made by dHPLC, SCD, thalassaemia, among other haemoglobinopathies, whether in the early development of the disease, or, for the determination of carriers. This research comprised different components (biochemical, haematological, and molecular), which will not only provide new knowledge for the approach and follow-up of SCD, but also a better understanding of the clinical manifestations of the disease.
Historical, geographical, and genetic context
Colombia is typical of South America’s genetic complexity: some of its regions (Chocó on the Pacific Coast just below Panama, or the coastal Caribbean populations, which include those found in Bolívar Department) exhibit a high proportion of African ancestry (Middleton et al. Citation2000; Bernal et al. Citation2010; Rosero and Bermúdez Citation2012; Noguera et al. Citation2014; Fong et al. Citation2013). This is in stark contrast to the high Native American gene pool in the southern and central inland regions of Colombia (Middleton et al. Citation2000; Díaz-Matallana and Martínez-Cruzado Citation2010; Yunis and Yunis Citation2013).
In the study of Noguera et al. (Citation2014), the results obtained reveal the presence of genetic substructure in the Bolívar department, attributable to a heterogeneous pattern of admixture. Overall, Europeans have contributed to more than 70% of the Y chromosomes in the current populations, except in Palenque where, consistent with historical data [from linguistic evidence (Schwegler Citation2012)], the genetic pool of African origin was highly preserved. The Caribbean populations, both insular and continental, are known to have a strong African ancestry. Although they found a predominant European contribution to the populations of Bolívar, in the Northern coastal region of Cartagena and surrounding areas, almost 25% of the Y chromosomes have sub-Saharan African ancestry.
The “Canal del Dique” is an artificial bifurcation of the Magdalena River, in the Caribbean Region of Colombia. It was built under orders from the Hispanic era in the sixteenth century to facilitate navigation between this river and Cartagena de Indias. It is 115 km long; it emerges from the Magdalena River at the height of Calamar and flows mainly into the Cartagena Bay (Aguilera-Díaz Citation2006). So, in 1650 the “Canal del Dique” was opened, which allowed fluvial communication with the Magdalena River, and the workforce included indigenous and Afro-descendants (Lucena-Giraldo Citation2017). In this study, several samples from the “Canal del Dique” Municipalities were analysed, which are located as follows: (1) Upper Canal del Dique: Calamar, Arroyohondo; (2) Middle Canal del Dique: Mahates, Maria la Baja; and (3) Lower Canal del Dique: María la Baja, Turbana, Turbaco, Cartagena (Alvarado-Ortega Citation2001; Coneo-Mendoza et al. Citation2014).
Palenque de San Basilio is a village located within the municipality of Mahates, Bolívar department. The Palenque, a black community in rural Colombia, have an oral history of fugitive African slaves founding a free village near Cartagena in the seventeenth century (Ansari-Pour et al. Citation2016). The Cultural Space of Palenque de San Basilio encompasses social, medical, and religious practices as well as musical and oral traditions, many of which have African roots. The palenquero is the only creole language in Latin America with a lexical Spanish basis and grammatical characteristics of Bantu languages. The language constitutes a vital factor in reinforcing social cohesion among community members (UNESCO Citation1992–2023). Recently, linguists identified 200 words, mainly spoken in the Congo region (Ansari-Pour et al. Citation2016).
In relation to the above, this “Canal del Dique” favoured miscegenation, as well as gene flow among said communities. Cartagena de Indias being one of the main seaports of Colombia in Northwestern South America, which introduced around 200,000 slaves from Africa, particularly Western and Central Western Africa, during the XV and XIX centuries to work in gold mines (Cuéllar-Ambrosi et al. Citation2000; Navarrete Citation2005).
From 1640 the Colombian Pacific coast received a massive influx of slaves from the West coast of Africa (Benin, Ivory Coast, Guinea-Bissau, Senegal, Sierra Leone, and Mali), mainly to work in the local mines (Fong et al. Citation2013).
The archipelago of San Andrés y Providencia (Colombia) was discovered by Christopher Columbus in 1492, and some Spaniards settled on the Santa Catalina Island, in 1641 the first African slaves arrived with new immigrants from England, in 1664 it was taken by the crew of the pirate Manswelt, in 1670 by the English corsair Morgan, and in 1689 it was again under Spanish rule. In 1806 it was occupied by English troops, and again it was under Spanish rule until 1886 when it was ceded to the central government (Bernal et al. Citation1995).
In the American continent, the presence of abnormal haemoglobins has been the product of both immigration and the interbreeding of various African and European populations with Amerindian groups, causing ethnic mixing with the introduction of mutations in the globin gene. Its distribution seems to follow a pattern closely related to the displacement that the populations of African origin have had in the national territory; the highest frequency is found in coastal populations, finding an abrupt decrease in populations along the Andes Mountain range (Varela et al. Citation2013). Consistent with Durán et al. (Citation2012), the distribution of haplotypes in the cities/municipalities included in this study suggests that individuals coming from central Africa (CAR) dispersed throughout the Colombian territory, and therefore this haplotype is more frequent in Colombia, followed by BEN – the Western Africa haplotype ( and ; and ).
Conclusions
This study is the first in Colombia that incorporates the use of SNP markers through Sanger’s DNA sequencing for the detection of SCD, as well as for the classification of the classic haplotypes associated with the β-globin cluster ( and Table S4; , , , and Figure S3 (a,b)), since the previously reported works on SCD were based on PCR-RFLPs markers (Bernal et al. Citation1995; Cuéllar-Ambrosi et al. Citation2000; Bernal et al. Citation2010; Alvear et al. Citation2012; Durán et al. Citation2012; Rosero and Bermúdez Citation2012; Fong et al. Citation2013; Ortega et al. Citation2015; Romero-Sánchez et al. Citation2015), or PCR-RFLPs and TaqMan procedure (Fong et al. Citation2015), and most of this scientific literature was previously reviewed by Díaz-Matallana et al. (Citation2021).
From the results of 101 samples analysed from Colombia in this work, the vast majority from the Department of Bolívar, the structural haemoglobinopathies HbAS (34.65%), HbSS (2.97%) and HbAC (1.98%) were found (, Table S4). Regarding the classical haplotypes analysed from 26 samples, mostly from the “Canal del Dique”, Bolívar, the haplotypes inferred by SNPs were detected in the order CAR (50.0%), BEN (30.76%), CAM (7.69%), ATP-I (7.69%) and SEN (3.84%), with CAR (Central Africa) being the most frequent haplotype in Colombia followed by the BEN haplotype (West Africa) (; and ), which is consistent with other studies (). The AI (Arabian Indian) haplotype was not detected in the samples analysed.
Our results evidenced the importance of SNPs identification by Sanger’s DNA sequencing to clarify confusing data, complement data and optimise the quality of diagnoses made by dHPLC of SCD and thalassaemia, among other haemoglobinopathies, whether in early development of the disease, or for the determination of carriers. This research included different components: biochemical, haematological, and molecular, which will not only provide new knowledge for the approach and monitoring of SCD, but also a better understanding of the clinical manifestations of the disease.
This research wants to highlight the role played by neonatal screening for haemoglobinopathies which, alongside an improved molecular diagnosis (SNPs identification by Sanger’s DNA sequencing), an education program and genetic counselling, would prevent the appearance of new cases after determining possible carriers or those affected, and therefore implications on offspring. Moreover it would allow early treatment of homozygous individuals, which would favour the survival of the patient and would significantly improve both their quality of life and that of their relatives. The main treatments for SCD were cited by Díaz-Matallana et al. (Citation2021).
In fact, nowadays gene therapies for both SCD and β-thalassaemia based on the CRISPR technique are a true reality. On November 16th 2023, marking a historic milestone, the United Kingdom became the first country to approve a gene-edited drug. The Casgevy treatment uses CRISPR to prevent debilitating pain in patients with SCD. It also eliminates the need for regular blood transfusions in people with β-thalassaemia (Wong Citation2023). On December 8th 2023, it was also approved in the United States for cases of SCD (FDA – U.S. Food and Drug Administration Citation2023). Lastly, on December 15th 2023, the EMA recommended approval of the first medicine using CRISPR/Cas9 (Casgevy), a novel gene-editing technology to treat β-thalassaemia and severe SCD in the European Union (EMA – European Medicines Agency Citation2023). Such treatment will open a hopeful path for these patients, however these gene therapies are very expensive at the moment, and it will be a challenge to make the treatment accessible globally.
Supplemental Material
Download PDF (1,018.1 KB)Acknowledgements
We are grateful to MINCIENCIAS, Universidad de La Sabana, Universidad del Sinú, and Pregen, Colombia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Abdalla Elsayed MEA, Mura M, Al Dhibi H, Schellini S, Malik R, Kozak I, Schatz P. 2019. Sickle cell retinopathy. A focused review. Graefes Arch Clin Exp Ophthalmol. 257(7):1–13. doi: 10.1007/s00417-019-04294-2.
- Adekile A, Akbulut-Jeradi N, Al Khaldi R, Fernandez MJ, Sukumaran J. 2021. Diagnosis of SCD and HBB haplotyping in the era of personalized medicine: role of next generation sequencing. J Pers Med. 11(6):454. doi: 10.3390/jpm11060454.
- Aghajani F, Mahdavi MR, Kosaryan M, Mahdavi M, Hamidi M, Jalali H. 2016. Identification of β-globin haplotypes linked to sickle haemoglobin (Hb S) alleles in Mazandaran province, Iran. Genes Genet Syst. 91(6):311–313. doi: 10.1266/ggs.16-00005.
- Aguilera-Díaz MM. 2006. El Canal del Dique y su subregión: una economía basada en la riqueza hídrica (PDF). Documentos de trabajo sobre economía regional. Banco de la República, CEER, Cartagena, p. 1692-3715. https://web.archive.org/web/20111105200007/http://www.banrep.gov.co/documentos/publicaciones/regional/documentos/DTSER-72%20%28VE%29.pdf.
- Alvarado-Ortega M, editor. 2001. Canal del Dique. Plan de restauración ambiental (1a etapa). Barranquilla: Uninorte, p. 12.
- Alvear C, Barboza M, Alayón AN, Viola M, Araque LM. 2012. Pilot study of haemoglobinopathies in newborns of the Rafael Calvo maternity clinic of Cartagena, Colombia. Colomb Med. 43:197–198.
- Ansari-Pour N, Moñino Y, Duque C, Gallego N, Bedoya G, Thomas MG, Bradman N. 2016. Palenque de San Basilio in Colombia: genetic data support an oral history of a paternal ancestry in Congo. Proc Biol Sci. 283(1827):20152980. doi: 10.1098/rspb.2015.2980.
- Bender MA. 2019. Sickle cell disease. Department of Pediatrics, University of Washington, Fred Hutchinson Cancer Research Center, Seattle, Washington (USA). National Organization for Rare Disorders (NORD), Inc. https://rarediseases.org/rarediseases/sickle-cell-disease.
- Bernal M, Collazos A, Bonilla RD, Tascón EP. 2010. Determination of the prevalence of haemoglobin S, C, D, and G in neonates from Buenaventura. Colomb Med. 41:141–147.
- Bernal MDP, Giraldo A, Bermudez AJ, Moreno E. 1995. Estudio de la frecuencia de hemoglobinopatías en las islas de San Andrés y Providencia. Biomedica. 15(1):5–9. doi: 10.7705/biomedica.v15i1.850.
- Centers for Disease Control and Prevention (CDC). 2015. Hemoglobinopathies: current practices for screening, confirmation and follow-up. Association of Public Health Laboratories, US Dept of Health, and Human Services. 57. p.
- Coneo-Mendoza Y, López-Pineda LF, Barraza-Pava ME, Sáenz-Zapata JA. 2014. La Subregión del Canal del Dique: un mar de Necesidades en un océano de riqueza. Colombia: CEDEC. Primera edición. ©Cámara de Comercio de Cartagena; p. 328.
- Cruz PRS, Ananina G, Gil-da-Silva-Lopes VL, Simioni M, Menaa F, Bezerra MAC, Domingos IF, Araújo AS, Pellegrino R, Hakonarson H, et al. 2019. Genetic comparison of sickle cell anaemia cohorts from Brazil and the United States reveals high levels of divergence. Sci Rep. 9(1):10896. doi: 10.1038/s41598-019-47313-2.
- Cuéllar-Ambrosi F, Mondragón MC, Figueroa M, Préhu C, Galactéros F, Ruiz-Linares A. 2000. Sickle cell anaemia and beta-globin gene cluster haplotypes in Colombia. Haemoglobin. 24(3):221–225. doi: 10.3109/03630260008997529.
- dbSNP Short Genetic Variations. 2022. National Center of Biotechnology Information (NCBI), U.S. National Library of Medicine. Available from: https://www.ncbi.nlm.nih.gov/SNP
- Díaz-Matallana M, Gómez Gutiérrez A, Briceño I, Rodríguez Cuenca JV. 2016. Genetic analysis of Paleo-Colombians from Nemocón, Cundinamarca provides insights on the early peopling of northwestern South America. Rev Acad Colomb Cienc Ex Fis Nat. 40(156):461–483. doi: 10.18257/raccefyn.328.
- Díaz-Matallana M, Márquez-Benítez Y, Martínez-Lozano JC, Briceño I, Benavides-Benítez E, Bernal JE. 2021. Anemia falciforme: una revisión sobre el genotipo de la enfermedad, haplotipos, diagnóstico y estudios asociados. Rev Méd Chile. 149(9):1322–1329. doi: 10.4067/S0034-98872021000901322.
- Díaz-Matallana M, Martínez-Cruzado JC. 2010. Estudios sobre ADN mitocondrial sugieren un linaje predominante en la Cordillera Oriental de Colombia y un vínculo suramericano para los arcaicos de Puerto Rico. Univ Med. 51(3):241–272. doi: 10.11144/Javeriana.umed51-3.esam.
- Díaz-Matallana M. 2015. Caracterización Genética de un grupo Paleoamericano Checua proveniente de Nemocón-Cundinamarca, Colombia: implicaciones para el Poblamiento Temprano de Suramérica [Tesis Ph.D]. Bogotá (DC): Ciencias Biológicas, Pontificia Universidad Javeriana.
- Durán CL, Morales OL, Echeverri SJ, Isaza M. 2012. Haplotipos del gen de la globina beta en portadores de hemoglobina S en Colombia. Biomédica. 32:103–111.
- El-Hazmi MAF, Warsy AS, Bashir N, Beshlawi A, Hussain IR, Temtamy S, Qubaili F. 1999. Haplotypes of the β-globin gene as prognostic factors in SCD. East Mediterr Health J. 5(6):1254–1258. doi: 10.26719/1999.5.6.1154.
- EMA – European Medicines Agency. 2023. First gene editing therapy to treat beta thalassemia and severe sickle cell disease [cited 2023 Dec 15]. www.ema.europa.eu.
- Fatima SS, Mussaed EM. 2015. Gene sequencing and SNP analysis of rs334 of beta haemoglobin involved in sickle cell anemia. Helix. 2:645–647.
- FDA – U.S. Food and Drug Administration. 2023. FDA approves first gene therapies to treat patients with sickle cell disease [cited 2023 Dec 8]. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease.
- Figueiredo MS, Silva MCBO, Guerreiro JF, Souza GP, Pires ACR, Zago MA. 1994. The heterogeneity of the βS cluster haplotypes in Brazil. Gene Geogr. 8:7–12.
- Fong C, Lizarralde-Iragorri MA, Rojas-Gallardo D, Barreto G. 2013. Frequency and origin of haplotypes associated with the beta-globin gene cluster in individuals with trait and sickle cell anemia in the Atlantic and Pacific coastal regions of Colombia. Genet Mol Biol. 36(4):494–497. doi: 10.1590/S1415-47572013000400005.
- Fong C, Menzel S, Lizarralde MA, Barreto G. 2015. Genetic variants associated with fetal hemoglobin levels show the diverse ethnic origin in Colombian patients with sickle cell anemia. Biomedica. 35(3):437–443. doi: 10.7705/biomedica.v35i3.2573.
- GE Healthcare. 2010. Whatman Application Note-28-9822-22AA.
- Gómez-Gutérrez A, Casas-Gómez C. 2014. Interpretación Clínica del Laboratorio. Bogotá: Editorial Médica Panamericana. p. 846.
- Gomez A, Kurf B. 2018. Chapter 5. Genetic testing techniques – Sanger sequencing. In: Robin NH, Farmer MB, editors. Pediatric cancer genetics. Canada: Elsevier; p. 47–64. https://www.sciencedirect.com/science/article/abs/pii/B9780323485555000053.
- Hanchard N, Elzein A, Trafford C, Rockett K, Pinder M, Jallow M, Harding R, Kwiatkowski D, McKenzie C. 2007. Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genet. 8(1):52. doi: 10.1186/1471-2156-8-52.
- Ibarra-Cortés B, Guzmán LF, Perea-Díaz FJ. 2019. Anexo IV. Anemia drepanocítica. In: del Castillo-Ruiz V, Uranga-Hernández RD, Zafra de la Rosa G. Genética Clínica. 2nd ed. México (DF): Manual Moderno; p. 651.
- Jiménez-Cobo C, Sebastián- Pérez E, Sevilla-Navarro J. 2021. Hemoglobinopatías: talasemia y drepanocitosis. Pediatría Integral. 25 (5):241.e1–241.e13.
- Joutovsky A, Hadzi-Nesic J, Nardi MA. 2004. HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: a study of 60,000 samples in a clinical diagnostic laboratory. Clin Chem. 50(10):1736–1747. doi: 10.1373/clinchem.2004.034991.
- Lettre G. 2012. The search for genetic modifiers of disease severity in the β-hemoglobinopathies. Cold Spring Harb Perspect Med. 2(10):a015032. doi: 10.1101/cshperspect.a015032.
- Lucena-Giraldo M. 2017. “Una obra digna de romanos”: El Canal del Dique, desde su apertura hasta la independencia. Expediente sobre el Canal del Dique (1647-63). Archivo General de Indias, AGI, Santa Fe, 199. https://www.banrepcultural.org/biblioteca-virtual/credencial-historia/numero-287/una-obra-digna-de-romanos-el-canal-del-dique-apertura-independencia.
- Lyra IM, Gonçalves MS, Pellegriinei JA, Gesteira M, Carvalho MH, Olalla ST, Figueiredo MS, Costa FF. 2005. Clinical, haematological, and molecular characterization of sickle cell anemia pediatric patients from two different cities in Brazil. Cad Saúde Pública. 21:1287–1290.
- MaxStat. 2015. MaxStat Lite v.3.60 program (Jever-OT Cleverns, Germany). Available from: https://maxstatlite.en.softonic.com/
- Medline Plus. 2023. Glucose-6-Phosphate dehydrogenase deficiency. Bethesda (MD): National Library of Medicine – NIH. Last Updated April 12, 2023. https://medlineplus.gov/genetics/condition/glucose-6-phosphate-dehydrogenase-deficiency/.
- Middleton D, Williams F, Meenagh A, Daar AS, Gorodezky C, Hammond M, Nascimento E, Briceno I, Perez MP. 2000. Analysis of the distribution of HLA-A alleles in populations from five continents. Hum Immunol. 61(10):1048–1052. doi: 10.1016/s0198-8859(00)00178-6.
- Moreno N, Martínez JA, Blanco Z, Osorio L, Hackshaw P. 2002. Beta-globin gene cluster haplotypes in Venezuelan sickle cell patients from the state of Aragua. Genet Mol Biol. 25(1):21–34. doi: 10.1590/S1415-47572002000100005.
- Muniz A, Corral L, Alaez C, Svarch E, Espinosa E, Carbonell N, di Leo R, Felicetti L, Nagel RL, Martinez G. 1995. Sickle cell anemia and beta-gene cluster haplotypes in Cuba. Am J Hematol. 49(2):163–164. doi: 10.1002/ajh.2830490212.
- Muñiz A, Puig A, Cabrera M, Fernández J, Martínez G. 2000. Marcadores genéticos en pacientes con anemia drepanocítica de la provincia de Cienfuegos: haplotipos del bloque α y β - talasemia. Rev Cubana Hematol Inmunol Hemoter. 16:142–144.
- National Center of Biotechnology Information (NCBI). 2022. dbSNP SHORT GENETIC VARIATIONS. U.S. National Library of Medicine. https://www.ncbi.nlm.nih.gov/SNP.
- Navarrete MC. 2005. Génesis y Desarrollo de la Esclavitud en Colombia: siglo XVI y XVII. Cali: Programa Editorial Universidad del Valle; p. 377.
- Noguera MC, Schwegler A, Gomes V, Briceño I, Alvarez L, Uricoechea D, Amorim A, Benavides E, Silvera C, Charris M, et al. 2014. Colombia’s racial crucible: Y chromosome evidence from six admixed communities in the Department of Bolivar. Ann Hum Biol. 41(5):453–459. doi: 10.3109/03014460.2013.852244.
- Old JM. 2003. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev. 17(1):43–53. doi: 10.1016/s0268-960x(02)00061-9.
- OMIM Database. 2022. Sickle cell anemia. Johns Hopkins University. https://www.omim.org/entry/603903?search=sickle%20cell%20anemia&highlight=cell%20anemia%20anaemia%20sickle
- OpenSNP. 2023. https://opensnp.org/.
- Orphanet. 2021. The portal for rare diseases and orphan drugs. Sickle cell anemia. https://www.orpha.net/consor/cgi-bin/index.php?lng=EN.
- Ortega DC, Fong C, Cárdenas H, Barreto G. 2015. Evidence of over-dominance for sickle cell trait in a population sample from Buenaventura, Colombia. Int J Genet Mol Biol. 7(1):1–7. doi: 10.5897/IJGMB2014.0106.
- Powars DR, Chan L, Schroeder WA. 1990. Beta S-gene-cluster haplotypes in sickle cell anemia: clinical implications. Am J Pediatr Hematol Oncol. 12(3):367–374. doi: 10.1097/00043426-199023000-00022.
- Rihet P, Flori L, Tall F, Traore AS, Fumoux F. 2004. Haemoglobin C is associated with reduced Plasmodium falciparum parasitemia and low risk of mild malaria attack. Hum Mol Genet. 13(1):1–6. doi: 10.1093/hmg/ddh002.
- Romero-Sánchez C, Gómez Gutiérrez A, Duarte Y, Amazo C, Manosalva C, Chila M L, Casas-Gómez MC, Briceño Balcázar I. 2015. Variantes de hemoglobina en una población con impresión diagnóstica positiva para hemoglobinopatías en Colombia. Rev Med Chil. 143(10):1260–1268. doi: 10.4067/S0034-98872015001000004.
- Rosero MJ, Bermúdez AJ. 2012. Análisis de hemoglobinopatías en regiones afrocolombianas usando muestras de sangre seca de cordón umbilical. Acta Médica Colombiana. 37(3):118–124.
- Ruiz-Morales AJ, Gómez-Restrepo C. 2015. Epidemiología Clínica, Investigación Clínica Aplicada. 2nd ed. Bogotá (DC): Editorial Médica Panamericana. p. 625.
- Rusanova I, Cossio G, Moreno B, Javier Perea F, De Borace RG, Perea M, Escames G, Acuña-Castroviejo D. 2011. β-globin gene cluster haplotypes in sickle cell patients from Panamá. Am J Hum Biol. 23(3):377–380. doi: 10.1002/ajhb.21148.
- Sambrook J, Russell DW. 2001. Molecular cloning A laboratory manual. Vol. 1. 3rd ed. New York: Cold Spring Harbor Laboratory Press.
- Schaefer GB, Thompson JN. 2016. Genética Médica, Un Enfoque Integrado. México (DF): McGraw-Hill Interamericana; p. 372.
- Schwegler A. 2012. Sobre el origen africano de la lengua criolla de Palenque (Colombia). Much augmented Spanish version of ‘‘Palenque(ro): the search for its African substrate’’, Schwegler 2011 above. In: Maglia G, Schwegler A, editors. Palenque (Colombia): oralidad, identidad y resistencia. Un enfoque interdisciplinario. Bogotá: Instituto Caro y Cuervo & Universidad Javeriana; p. 107–179.
- Shaikho EM, Farrell JJ, Alsultan A, Qutub H, Al-Ali AK, Figueiredo MS, Chui DHK, Farrer LA, Murphy GJ, Mostoslavsky G, et al. 2017. A phased SNP-based classification of sickle cell anemia HBB haplotypes. BMC Genomics. 18(1):608. doi: 10.1186/s12864-017-4013-y.
- Shriner D, Rotimi C. 2018. Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the holocene wet phase. Am J Hum Genet. 102(4):547–556. doi: 10.1016/j.ajhg.2018.02.003.
- SNPedia. 2022–2023. https://www.snpedia.com/
- Thein SL, Menzel S. 2009. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol. 145(4):455–467. doi: 10.1111/j.1365-2141.2009.07650.x.
- Tripathi P. 2022. Genetics of thalassemia. In: Rajashekaraiah V. The erythrocyte – A unique cell. IntechOpen. doi: 10.5772/intechopen.106748.
- UNESCO. 1992–2023. Cultural Space of Palenque de San Basilio. https://ich.unesco.org/en/RL/cultural-space-of-palenque-de-san-basilio-00102.
- Varela I, Sequera A, Olivero R. 2013. Detección de hemoglobinopatías en recién nacidos del Hospital Materno Infantil “Dr. José María Vargas” de la ciudad de Valencia, Venezuela. Salud Online. 17(2):6–12.
- Wong C. 2023. UK is first nation to approve CRISPR therapy for diseases. Landmark decision could transform the treatment of sickle-cell disease and β-Thalassaemia. Nature. 623(7988):676–677. 23/11/2023). doi: 10.1038/d41586-023-03590-6.
- Yunis JJ, Yunis EJ. 2013. Mitochondrial (mtDNA) haplogroups in 1526 unrelated individuals from 11 Departments of Colombia. Genet Mol Biol. 36(3):329–335. doi: 10.1590/S1415-47572013000300005.