
Abstract
Lipid-based drug delivery systems (LBDDS) are one of the most studied bioavailability enhancement technologies and are utilized in a number of U.S. Food and Drug Administration (FDA) approved drugs. While researchers have used several general rules of thumb to predict which compounds are likely to benefit from LBDDS, formulation of lipid systems is primarily an empiric endeavor. One of the challenges is that these rules of thumb focus in different areas and are used independently of each other. The Developability Classification System attempts to link physicochemical characteristics with possible formulation strategies. Although it provides a starting point, the formulator still has to empirically develop the formulation. This article provides a review and quantitative analysis of the molecular properties of these approved drugs formulated as lipid systems and starts to build an approach that provides more directed guidance on which type of lipid system is likely to be the best for a particular drug molecule.
Introduction
An increasing percentage of drug molecules in pharma development pipelines can be classified according to the Biopharmaceutics Classification System (BCS) as Class II compounds (compounds having good permeability but poor solubility). Additionally, a considerable percentage of today’s pipeline molecules are both poorly soluble and poorly permeable (BCS Class IV). These newer drug molecules are discovered and optimized through relatively novel technologies such as high-throughput screening, fragment-based drug discovery, or computational modeling. While, they demonstrate superior potencies and specificities compared to their predecessors, many also possess inherent challenges in terms of oral delivery [Citation1]. The low solubility and poor permeability of these compounds translates into suboptimal patient outcomes due to poor oral bioavailability and variable pharmacokinetics.
Simple formulation approaches (conventional tablets or powder in capsule) are not enough to address these issues. Bioavailability enhancement technologies (LBDDS, solid amorphous dispersions, API salt formation, or API particle size reduction) have been developed to address the issue of low solubility by improving the dissolution rate and/or the apparent solubility of these drug molecules. A molecule’s dissolution rate can be enhanced by increasing the surface area (particle size reduction) or by stabilizing the amorphous form of the drug in a polymer (hot melt extrusion and spray drying). Lipid excipients and surfactants can be used to present and maintain the drug molecule in solution form both pre- and post-administration. Of the bioavailability enhancing technologies, LBDDS are the most well studied and well established as a formulation approach for addressing solubility and/or permeability issues [Citation2]. Therefore, we focus our efforts in this review on LBDDS.
There are numerous ‘Rules’ and ‘Classification Systems’ available to formulation scientists for prediction of in vivo performance. Formulators are working to create in silico tools and using classification systems coupled with in vitro dissolution tests in biorelevant media for predicting in vivo outcomes during the formulation design and screening phases of product development. While predicting the self-emulsification behavior of LBDDS excipient combinations is reasonable, formulators lack the in silico tools to accurately predict how specific drugs will behave in specific formulations [Citation3]. The aim of this study was to analyze the physicochemical characteristics of these drugs and classify them according to the different systems to see what, if any, correlations exist between physicochemical properties and classification systems or between classification systems. Furthermore, this exercise could validate the belief that simple physicochemical properties are insufficient for de novo LBDDS formulation design and highlight key areas for the focus of future studies.
We first examined U.S. Food and Drug Administration (FDA) approved drugs (based on NDA approvals) that have been formulated using LBDDS. We focused only on approved drugs with the rationale that they were robust enough to progress through all phases of drug development and receive FDA approval. Next, we classified the drugs using the Lipid Formulation Classification System (LFCS). We calculated physicochemical properties that may influence the choice of LBDDS used for formulation and used these properties to group the drugs into the BCS and Developability Classification System (DCS). Based on the these calculations and classifications, we sought any correlations that would provide guidance as to what types of drug molecules are good fit for LBDDS and what specific type of LBDDS. We conclude by proposing therapeutic categories, compound categories, and other untapped areas that can benefit from the advantages offered through the use of LBDDS.
FDA approved orally administered products that use lipid formulations
The increasingly overall water-insoluble nature of drug candidates has been accompanied by the increased use of solubilization technologies for FDA approved drugs. A recent analysis revealed that ∼6% of all approved new molecular entities from 1975 to 2013 utilized solubilization technologies [Citation2]. The same analysis showed that LBDDS are the most widely used solubilization technology. An analysis by Strickley in 2007 estimated that LBDDS oral drug products account for 2–4% of all commercially available drug products [Citation4].
A review of the literature and drug databases (DailyMed, DrugBank, and Drugs@FDA) revealed 27 unique drug molecules that were FDA approved as 36 different oral LBDDS. () [Citation5–7]. A few of the approved drugs have been discontinued. According to the Federal Register, none have been for safety or efficacy reasons. The three HIV protease inhibitor formulations were replaced by newer formulations. Saquanavir (Fortovase®) was discontinued because a film-coated tablet dosage form with a lower pill burden was introduced. Ritonavir/lopinavir (Kaletra®) was replaced by a solid dispersion formulation, which does not require refrigeration, had higher drug loading per dosage unit, and reduced daily pill burden. Amprenavir (Agenerase®) was succeeded by a pro-drug, fosamprenavir.
Table 1. FDA approved drugs utilizing lipid systems.
Analysis of these approved products can offer insights to those physicochemical characteristics of a drug molecule that make LBDDS an attractive formulation option. As seen in and , there is a wide diversity, in terms of pharmacological class and chemical structures, of approved drugs that have been formulated using lipid systems. In short, LBDDS are a versatile platform which can benefit a wide range of drug molecules. One reason for the versatility is the range of excipients used in LBDDS. The major categories of LBDDS excipients are lipids, water insoluble surfactants (HLB <12), water soluble surfactants (HLB >12), and hydrophilic cosolvents. The choice and relative percentage of excipients in LBDDS influence drug solubility, LBDDS dispersability, and formulation properties. This in part explains its historic and current popularity for formulating poorly soluble and/or permeable drugs.
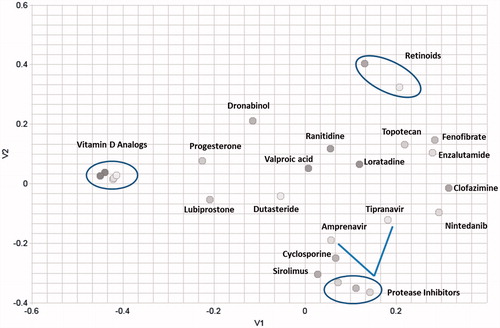
Figure 1. Multidimensional (2D) scaling of the chemical structures of the dataset reveals high structural diversity. The axes (V1 and V2) represent chemical space in two directions.
The interest and growth in lipid based systems can be traced to several findings and developments. Unless they are in solution following oral administration, poorly soluble drugs have poor absorption, variable pharmacokinetics, and tend to experience significant positive food effect. Lipid formulations partially improve the solubility of drugs by stimulating the same physiologic events, although to a lower magnitude, as administration of drugs with a fatty meal [Citation8].
The soft capsule has become the most common dosage form for the administration of lipid formulations (). Soft capsules are the utilized dosage form for 25 of 36 lipid formulations, 6 products are hard shell capsules, and 5 are oral solutions, The advantages of soft capsules have been extensively documented elsewhere [Citation9,Citation10]. The introduction of Neoral® in 1995 is a widely cited example illustrating how a self-emulsifying system (the first IIIA LFCS product) can address clinical concerns and result in a product providing a better treatment for patients. There were several approved LBDDS on the market prior to 1995 (NDA Approval Date of Neoral) and many more that followed (). Early lipid formulations consisted of pure oils (LFCS Class I and some Class II).
Designing lipid formulations
Unlike other formulation approaches, the absorption process of LBDDS is complex and dynamic. The formulation design considerations have clear and direct implications on drug absorption in vivo. The formulator needs to possess knowledge about physical chemistry, thermodynamics, and gastrointestinal physiology to ensure that drug precipitation does not occur in the gastrointestinal tract following dispersion and/or digestion of the formulation. While most LBDSS are designed to solubilize the drug in the formulation prior to administration, endogenously, the drug in the formulation matrix experiences environmental transition from a solution state in formulation, to an emulsified state due to dispersion upon initial contact with GI fluid, and finally a mixed micellar phase after lipid digestion.
LBDDS is an umbrella term for numerous delivery systems [oil solutions, emulsions, dispersions, micelles, self-emulsifying drug delivery systems (SEDDS), and self-microemulsifying drug delivery systems (SMEDDS)] and can be broadly classified into four types according to the LFCS proposed by Pouton () [Citation11,Citation12]. In our review, there were 16 drugs that utilized Type I LBDDS, 3 drugs formulated as Type II, 5 drugs as Type III, and 2 drugs as TYPE IV (). Type I lipid formulations contain oils that need to be digested in order to form mixed micelles. Type I formulations are typically biocompatible and simple (thereby explaining their early use), and often the LFCS formulation type first attempted if the dose of API can be fully solubilized in the fill. The vitamin D analogs and retinoids are formulated as Type I LBDDS LFCS Type II lipid formulations consist of mixtures of lipids and water- insoluble surfactants (HLB <12). They self-emulsify into crude oil-in-water (o/w) emulsions usually with energy input. LFCS Type III lipid formulations contain water- soluble surfactants (HLB >12) and co-solvents. These formulations spontaneously self-emulsify upon contact with an aqueous environment. They are commonly referred to as self-emulsifying drug delivery systems (SEDDS) when dispersion droplet size is >200 nm or as self-micro emulsifying drug delivery systems (SMEDDS) when dispersion droplet size is <200 nm. Finally, LFCS Type IV lipid formulations do not contain oil and are based on water-soluble surfactants and co-solvents. The fine dispersions formed when in contact with an aqueous environment translates to rapid drug release and absorption. The solvent capacity of these systems for maintaining a poorly soluble compound is usually limited which often results in the precipitation of the compound in the GI tract.
Table 2. Lipid formulation classification system.
Excipient use in LBDDS
LBDDS include a wide range of excipients. Some considerations that a formulator must consider include: regulatory acceptance, safety, solvent capacity, dispersion characteristics (usually in biorelevent media), digestability, interactions with efflux and metabolism processes (P-gp and CYP), stability, purity and amount/type of impurities, compatibility with the capsule, and cost. Excipient safety is a major consideration since the vast majority of FDA approved drugs using a LBDDS are intended for chronic use.
Lipids
By definition, LBDDS typically contain drug dissolved or suspended in oils (triglycerides or mixed glycerides). Of the 36 FDA approved formulations, 8 formulations did not contain oils and were classified as Classes III or IV according to the LFCS. Early lipid formulated drugs contain almost exclusively oils. Ritonavir (NDA approval: 1996) was the first LBDDS formulated drug that did not contain oil (). The biggest advantage of using oil is that lipophilic drugs are solubilized and dissolution of the drug within the GI tract is not required. Once digested, there are higher odds that the drug will stay in solution in vivo due to the micellization process. Oils can be divided into long chain triglycerides (LCT) and medium chain triglycerides (MCT) [Citation9]. Formulations containing these typically require digestion before absorption. Lipid digestion or lipolysis converts triglycerides into mono- and di-glycerides and fatty acids. The incorporation of mixed glycerides (mono- and di-glycerides and free fatty acids) in a formulation can improve the solvent capacity of lipid formulation in cases of compounds that have low solubility in triglycerides alone. Another advantage of using mixed glycerides is that they are similar to lipid digestion products. Undigested triglycerides have poor miscibility in the aqueous environment of the gastrointestinal tract, which leads to variation in gastric emptying and absorption [Citation13]. Corn oil was the oil component of cyclosporine (Sandimmune®) capsules (). The improved formulation (Neoral®) substituted corn mono- and di-glycerides for corn oil. The clinical result was less food effect and improved pharmacokinetic uniformity [Citation14,Citation15].
Surfactants and co-solvents
Surfactants are commonly used in lipid formulations to enhance drug solubility and self-emulsifying properties of the vehicle itself to minimize dependence on a patient’s digestion factors. Generally, nonionic surfactants have lower toxicity compared to ionic surfactants; therefore, LBDDS usually include nonionic surfactants. The marketed protease inhibitors, with the exception of saquinavir, contain considerable amounts of surfactants. Amprenavir capsules and tipranavir oral solution used vitamin E TPGS, which can behave as both a solubilizer and permeation enhancer. Ritonavir, ritonavir/lopinavir, and tipranavir capsules use polyoxyl 35 castor oil as the surfactant. Polyoxyl 35 castor oil is used to improve solubilization, but also has been shown to modulate P-glycoprotein (P-gp) activity on the apical membrane of the intestine and improve absorption of lipophilic drugs [Citation16]. P-gp on the intestinal villi is known to limit oral absorption of peptidomimetics such as protease inhibitors [Citation17]. Therefore, polyoxyl 35 castor oil can serve a dual purpose in formulation of drugs that are substrates for P-gp. As an example, early studies showed that saquinavir is a P-gp substrate and its oral bioavailability may be limited by P-gp efflux [Citation18]. However, the formulation does not contain any excipients that have the potential to inhibit drug efflux. More recent studies using digoxin as a probe in healthy subjects have shown that both saquinavir and ritonavir have inherent potential to inhibit P-gp [Citation19]. In some studies, ritonavir has also been demonstrated to induce P-gp activity [Citation20], suggesting a complex interaction of the drug and transporter which is still not completely understood.
Co-solvents such as propylene glycol and ethanol are used to increase drug solubilization but also serve to enhance the dispersion rate of lipid formulations and are often included in SEDDS and SMEDDS. However, upon dispersion in the GI fluids, they rapidly partition into the aqueous phase and reduce the solvent capacity of the formulation often leading to drug precipitation.
Physicochemical properties of lipid-formulated drugs
Choosing an appropriate formulation approach whether it be a lipid formulation or another type of formulation is typically an empiric endeavor that consumes significant time and resources (including API quantity). Because of limited API quantity at early stages, many scientists forego lipid formulation evaluation at drug discovery stages and instead utilize more conventional formulation approaches such as aqueous suspensions and powder in a capsule [Citation21]. The Lipid Formulation Classification System was developed to be used in conjunction with in vitro tests to create a system for the standardization of LBDDS which serves as a database correlating formulation composition to predicted in vitro–in vivo performance [Citation11]. During the development of a lipid formulation such as SEDDS, the formulator must take into account many considerations regarding the formulation strategy including excipients, solubility, stability, and scale-up and production [Citation22]. In general, the key design criteria for development of LBDDS are drug solubilization and prevention of drug precipitation in vivo. Beyond this, there exists the need for practical guidelines based on a database of molecules’ physiochemical properties to assist the formulator in predicting the most promising formulation pathways for a given drug compound. There are a couple of general rules based on experience. Pouton and Porter [Citation23] state that drugs with a LogP ∼2 and high melting points (numerically undefined) are usually poor candidates for lipid system and better suited for nanomilling or amorphous formulations. Lipid systems are excellent vehicles for highly lipophilic drugs (LogP >5). However, there are insufficient studies, particularly comparative studies, for the large portion of drugs that fall between these two extremes in LogP (drugs with LogP between 2 and 5). Similarly, Chen et al. [Citation21] proposed the following drug profile required for lipid formulations: aqueous solubility <10 mcg/ml; LogP >5; solubility in oils and lipids >25 mg/ml; relative low melting point; and good chemical stability.
Therefore, based on the need for additional guidelines for formulator’s considering a LBDDS approach, we sought to retrospectively analyze any commonalities or outliers in the set of FDA approved drugs using a LBDDS to address this need. We constructed box-and-whisker plots for the different physicochemical properties to illustrate the range, quartiles, and median values for each parameter. Restricting our analysis to this set of data provides some degree of assurance that these formulations are robust and have withstood the rigors of in vitro, animal, and human testing. The major drawback to this approach is that the dataset reviewed is somewhat limited in terms of number of drugs.
Chemical structure similarity of FDA approved drugs in LBDDS
Using an online program developed by the Girke lab, ChemMine Tools, we performed a multidimensional scaling of the drug molecules as shown in [Citation24]. The chemical structures of the drugs were entered into ChemMine Tools online platform and the dissimilarity analysis was performed with a similarity cutoff of 0.7. This approach clusters compounds by structural similarity. The program generates atom pair descriptors that are used to calculate a similarity matrix based on common and unique atom pairs among the compound set using the Tanimoto coefficient, which is the proportion of shared features between two compounds divided by the sum of common and unique features. The Tanimoto coefficient can range from 0 to 1 with a higher value means greater similarity. The distance matrix is calculated using an all-against-all comparison based on atom pair similarity and translating the similarity scores into distance values. We assessed the chemical structures in a two dimensional plane. The axes V1 and V2 are the axes defining two directions in a plane. As seen in , the molecules are quite diverse in structure. The axes represent molecular spaces in two dimensions. The most common drug classes (circled in ) are vitamin D analogs, protease inhibitors, and retinoids. These classes occupy different regions of the graph. The five vitamin D analogs are distinct from any other molecule structures and located on the left side of the graph. The retinoids are distinct from the other molecules as well and located on the upper right. The protease inhibitors occupy space on the bottom right. Tipranvair is the only non-peptidomimetic protease inhibitor and is evidenced as slightly outside of the cluster of the other protease inhibitors. The protease inhibitors are proximate to sirolimus and cyclosporine. The similarity is likely due to the peptidomimetic nature of protease inhibitors. This mapping illustrates that LBDDS is applicable to a wide range of molecular structures.
Molecular weight of FDA approved drugs in LBDDS
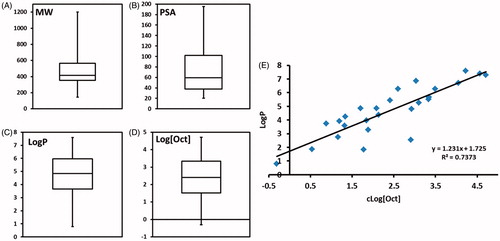
The molecules in exhibit a wide range of molecular weights from 144 Daltons (Da) for valproic acid to 1202.64 Da for cyclosporine with the median molecular weight of 416.64 Da (). A large portion of these drugs have ‘high’ molecular weights; 9 of the 27 drug molecules have molecular weights above 500 Da. This group of high molecular weight drugs is comprised of the protease inhibitors, cyclosporine A, dutasteride, nintedanib, and sirolimus (Supplementary Table S1).
Figure 2. Quantitative assessment of chemical properties. (A) molecular weight, (B) polar surface area, (C) LogP, (D) logarithm of octanol solubility, and (E) correlation between octanol solubility and LogP.
Permeability profiles of FDA approved drugs in LBDDS
There is evidence that the components of LBDDS impact intestinal permeability by changing the passive permeability and/or by inhibiting efflux transport [Citation25–28]. Medium chain fatty acids, lysophospholipids, surfactants, and cosurfactants increase passive permeability via tight junction opening or via increased membrane solubilization and increased membrane fluidity [Citation29]. We sought to evaluate drug molecules in our dataset to see which ones had the physicochemical characteristics indicative of poor permeability and if they were formulated with excipients that have been shown to improve permeability. A study of over 1100 GlaxoSmithKline drug candidates revealed that < 10 rotatable bonds and a polar surface area (PSA) < 140 Å display better permeation rates [Citation30]. Another study showed that in addition to PSA, LogD at pH =7.4 and radius of gyration influence Caco-2 permeation [Citation31]. In our analysis, we looked at the PSA, which is the summation of the surface area of all polar atoms (oxygen and nitrogen) and their attached hydrogens. We also calculated the permeability in human jejunum at pH 6.5 using the ACD The median PSA of the dataset is 59.45 Å () indicating that overall these molecules have very good permeability properties. In general, the protease inhibitors have relatively high PSAs (>100 Å). These molecules also have high molecular weights (> 500 Da). Only three drugs (ritonavir, saquinavir, and sirolimus) in our dataset have a PSA above 140 Å (Supplementary Table S1). Ritonavir (Norvir®) is a LFCS Type III LBDDS and consists of oleic acid, polyoxyl 35 castor oil, and ethanol. Saquinavir (Fortovase®) is a LFCS Type I LBDDS and contains only medium chain mono- and di-glycerides. Sirolimus (Rapamune®) was formulated as a LFCS Type III LBDDS using phosphatidylcholine, mono- and di-glycerides, soy fatty acids, ascorbyl palmitate, polysorbate 80, ethanol, and propylene glycol. Polyoxyl 35 castor oil and polysorbate 80 have been shown to modulate P-glycoprotein efflux pump and effects on phospholipid membranes [Citation32,Citation33]. Good permeation is a prerequisite for molecules that exert their pharmacological effect via nuclear receptor binding (retinoids, vitamin D analogs, and progesterone) or need to cross the blood-brain barrier (dronabinol or valproic acid). It is no surprise that these molecules have the lowest PSAs (PSA range: 20.23–60.69 Å).
Solubility profiles of FDA approved drugs in LBDDS
Poorly water soluble drugs represent a diverse set of compounds with different physicochemical properties. Most scientists believe that lipid systems are designed for highly lipophilic drugs (LogP >2). Broadly speaking, poorly water soluble drugs can be classified as non-lipophilic hydrophobic (brick dust) or lipophilic hydrophobic (grease balls) [Citation34]. ‘Brick dust’ compounds have tight crystal lattices (and higher melting temperatures), are not soluble in glyceride excipients, and require formulation with surfactants or co-solvents (LFCS Types IIIB and IV). On the other hand, ‘grease balls’ have good solubility in lipid excipients and can be formulated into LBDDS (LFCS Types I, II, and IIIA).
Our analysis showed the dataset of approved drugs in primarily consisted of lipophilic compounds with a median LogP of 4.85 and an interquartile range of 3.66–5.97 (). This analysis reveals a significantly higher LogP profile than most approved drugs based on an analysis by Leeson and Springthrope looking at LogP and molecular mass trends for pipeline and approved drugs [Citation35]. In their year-by-year analysis of 592 drugs launched between 1983 and 2007, the median cLogP for launched drugs failed to eclipse a cLogP of 4 for any year. With one exception, the median cLogP for every year fell between cLogP of 2 and 4. There was weak correlation of increasing median cLogP with each passing year and the slope of the straight fit line was very small. A commonly held belief is that drugs with a LogP >2 [23], are good candidates to be formulated as LBDDS. Based on the analysis by Leeson and Springthrope, nearly half of drug candidates should be formulated as LBDDS. The LogP >2 rule of thumb is not sufficiently discriminatory and sets the LogP bar too low. Our analysis showed that the median LogP of 4.85 for lipid formulated drugs. Amprenavir, ranitidine, and topotecan were the only compounds with a LogP below 2 (Supplementary Table S1) and are likely outliers. The molecules with the next lowest LogP values were valproic acid and lubiprostone with values of 2.54 and 2.76, respectively. This suggests that using a LogP of 2 to filter molecules that benefit from lipid formulation may be setting the level too low. A better approach to decide whether a drug is a good candidate to be formulated as a LBDDS is to look at other factors as done with the DCS.
We set to further analyze the data set’s LogP values. Our goal was to determine if LogP value is a sufficient parameter to determine whether a compound has suitable solubility in lipid excipients to be formulated in a lipid system. A compound’s LogP is a partition coefficient and influenced by two variables: its solubility in octanol and its solubility in water. We sought to assess the relative contribution of each variable on the LogP. In other words, are higher LogP driven by increasing lipid solubility, decreasing water solubility, or both? Using predicted LogP and predicted water solubility values, we were able to calculate the octanol solubility of FDA approved drugs formulated in lipid systems (). The calculated octanol solubility served as a surrogate for sufficient solubility in lipid excipients/lipid formulation solvent capacity. The water content in lipid formulation is absent or negligible. Therefore, we sought to assess if drug solubility in octanol has value as a predictor whether a lipid formulation is the best option compared to LogP. While this is true, LogP may be a better measure of consequences of LBDDS in vivo. This approach aims to minimize substantial influence of poor water solubility on LogP prediction. From , it is seen that the logarithm of the octanol solubility (Log[Oct]) has a linear relationship with the predicted LogP. The slope and y-intercept above 1 indicate that overall, the compounds in the dataset have a lipophilic tendency.
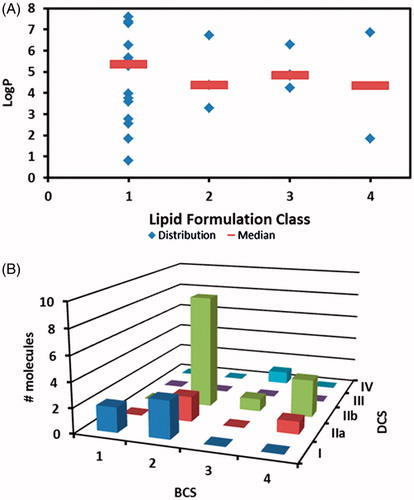
Based on our dataset (), there is no preference for lipid formulation class (LFCS) based on a molecule’s LogP value. Type I formulations, which are pure oils, have been used for drugs with a wide range of LogP values. It is surprising that Type I lipid formulation have been developed for hydrophilic molecules. The formulation rationale is not apparent.
Figure 3. Integration of different classification systems. (A) applicability of lipid formulation based on compound’s LogP. Markers represent individual LogP and line represents median LogP (B) relationship between BCS and DCS.
To support further formulation development studies, formulators try to target drug solubility of 25–50 mg/ml (log scale: 1.4–1.7) in lipid excipients [Citation21]. Our calculations show that 7 drugs have octanol solubility below this rule of thumb. Lubiprostone is a LFCS Type I, ranitidine is LFCS Type IV, and the rest fall into LFCS Type IV. Because lipid formulations are multicomponent systems and have complex behavior, it is difficult to create an in-silico prediction for determining if a drug has sufficient solubility in lipid systems for further formulation development.
Enzalutamide (LogP 3.75, log[Oct] 0.88) has both poor aqueous and octanol solubility, which combine to demonstrate a rather high LogP. Ranitidine, topotecan and amprenavir had the lowest predicted LogP (ALOGPS) at 0.79, 1.84, and 1.85, respectively. Amprenavir (72–74 °C) [Citation36] and ranitidine (69–70 °C) have low melting points. Topotecan has a melting point of 213–218 °C .Based on the above mentioned general rule, topotecan would be better formulated as an amorphous solid dispersion.
Classification according to BCS and DCS
The BCS was proposed by Amidon and colleagues to correlate in vitro dissolution and in vivo bioavailability and illustrate that solubility and permeability were important to oral drug absorption [Citation37]. Drugs fall within one of four classes based on their permeability and solubility. The majority of lipid formulated drugs fall into either BCS Class 2 (good permeability, poor solubility) or Class 4 (poor solubility and permeability). The four exceptions in our dataset are ergocalciferol (BCS Class 3), valproic acid (BCS Class 1), ranitidine (BCS Class 3), and topotecan (BCS Class 1).
While the BCS is a good regulatory system for identifying drugs that may be candidates for biowaivers based on in vitro dissolution data, it is not very useful for formulators to determine suitable formulation approaches. Many of the problematic compounds fall within the BCS Class 2, but there is insufficient knowledge of their solubility behavior. The Developability Classification System (DCS) was developed by Butler and Dressman to be used as a formulation tool [Citation38]. The DCS makes a couple of important amendments to the BCS. The solubility of the highest strength dose unit is replaced by the highest single dose, the medium for solubility determination is changed from buffer to fast state simulated intestinal fluid, and the fluid volume is for solubility determination is increased from 250 to 500 ml to be more representative of in vivo. The permeability and solubility characteristics (good or poor) for each of the four classes align between the BCS and DCS. Like Class 1 BCS drugs, DCS Class 1 drugs have good solubility and permeability. Drugs falling into Class 2 DCS have good permeability and poor solubility. They are subdivided Classes IIa and IIb based on their solubility limited absorbable dose. The absorption of drugs in DCS Class IIa is considered to be dissolution rate-limited because the dissolution rate is too slow for all drug particles to be in solution at the absorption site. The absorption of Class DCS IIb drugs is solubility-limited, which means that there is insufficient GI tract fluid to dissolve the dose. DCS Class III drugs have good solubility and poor permeability and those in DCS Class IV have poor permeability and solubility.
Classification of compounds in the dataset according to the DCS resulted in the molecules being placed in the following DCS Classes: Class I (n = 6), Class IIa (n = 5), IIb (n = 15), III (n = 0), and IV (n = 1) (). The dose/solubility ratio was calculated and plotted on the x-axis. A higher value indicated poorer solubility in fasting state simulated intestinal fluid. The calculated effective permeability in human jejunum is plotted on the y-axis. As expected, the majority of drug molecules fall within DCS Class IIb, those compounds with intrinsic solubility limitations. It is interesting to note the relatively high percentage of drugs that were classified as DCS Class I. Ranitidine was the only drug with poor permeability (< 1 × 10−4 cm/sec). As seen in , most BCS Class 2 drugs are DCS IIb with several falling into DCS Class I and IIa. The majority of drugs in BCS Classes 3 or 4 are typically DCS Class IIb compounds with a few in DCS Classes IIa and IV.
Figure 4. DCS plot of FDA approved drugs formulated using LBDDS with LFCS represented by different markers.
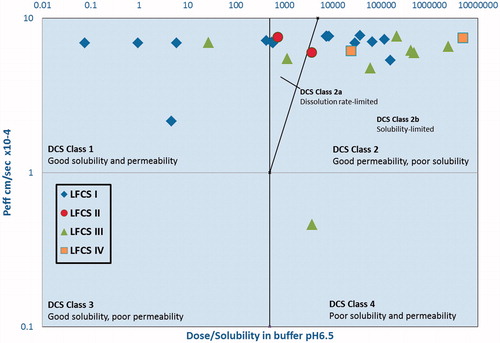
We sought to evaluate whether DCS class could aid formulators to choose the most appropriate lipid formulation type (LFCS) for solubilization of the entire drug dose and prevention drug precipitation in vivo. In the DCS plot (), we indicated the LFCS Class using different shapes for each drug. We had expected that Type I LBDDS, which consist of pure oils, would be best suited for the most poorly soluble drugs (those with the highest dose/solubility ratio). However, there were several DCS Classes 1 and 2a drugs formulated as Type 1 LBDDS. There are numerous factors in addition to excipient type. Fatty acid chain length of lipid excipients is important as it influences lipid digestion and drug solubilization. Less lipophilic drugs that can be easily solubilized to the aqueous GI environment can be best formulated with medium chain glycerides [Citation39]. Highly lipophilic drugs are better formulated using long chain glycerides in order that these drugs remain in the undigested oil phase or partition into mixed micelles following in vivo administration [Citation40]. Furthermore to plotting the drugs on the DCS plot, we sought to examine if drugs that fall within a certain region of the DCS plot will be better formulated as a self-emulsifying system (LFCS IIIA or IIIB). From our analysis (), there is no clear correlation between DCS Class and LFCS formulation type, and in fact the data is in some cases counter-intuitive. This is the likely due to the limited dataset. As shown is , the majority of molecules falling into DCS class I appear to be formulated as Type I lipid formulations. Type I lipid formulations were used for the DCS classes I, IIa, and IIb (). DCS IIb compounds, which were the most common, were shown to be formulated using all types of lipid systems with a preference for Type I lipid formulations.
FDA approved molecules in LBDDS beyond rule of five
Lipinski’s Rule of Five (Ro5) is a rule of thumb used during drug development to evaluate drug-likeness of a molecule or determine if a molecule is likely to be orally active in humans [Citation41]. The Ro5 describes chemical properties important for pharmacokinetics and does not predict pharmacodynamics. There are four criteria: ≤5 hydrogen bond donors (HBD); ≤ 10 hydrogen bond acceptors (HBA); molecular weight (MW) < 500 Daltons; and LogP <5. The Rule is said to be ‘broken’ when 2 criteria are not met. Adhering to the strict guideline, most commercial oral drugs do not violate the Ro5. We evaluated the list of FDA approved drugs formulated as lipid systems to see if any drugs violated the Ro5 and found 5 cases:
Sirolimus (MW 914.17, LogP 7.45, HBA 12)
Saquinavir (MW 670.84, HBD 6)
Tipranavir (MW 602.7, LogP 6.29)
Dutasteride (MW 528.50, LogP 5.45)
Cyclosporine (MW 1202.64, HBA 16)
It is rather widely accepted that target biology and chemical properties influences the physicochemical properties of ligands. These targets tend to have lipophilic or large, flat binding pockets. An analysis of recently patented molecules, Bergström et al. [Citation42] discovered that molecules are almost exclusively lipophilic for certain targets. Approximately 50% of the targets had molecules with an average LogP ≥4. With a limited dataset of approved drugs using LBDDS, we were unable to extend our analysis across a target or drug class. Our analysis revealed that 64% (16 of 25) drug molecules had a LogP >4. Of the molecules violating the Ro5, three compounds have high LogP values.
An analysis by Paul Leeson showed that while increasing lipophilic character of drugs began after 1990 (predicted LogP of ∼2.5 from 1950–1990, predicted LogP ∼3.3 since 1990), there are been a steady increase in molecular weights since the 1950s (MW ∼300 in the 1950s to ∼450 in the 2000s) [Citation43]. In our dataset, molecular weight >500 is the most common physicochemical property violation for molecule breaking the Ro5. Our review revealed that 9 of 25 molecules had molecular weights >500. Protease inhibitors are known for the high molecular weights, but tipranavir is only protease inhibitor with a LogP that violates the Lipinski parameter. This may be explained based on chemical structure since tipranavir does not contain a peptidomimetic hydroxyethylene core and other protease inhibitors do.
LBDDS to reduce magnitude of food effect
The absorption of poorly water soluble drugs is improved when administered with food, termed positive food effect [Citation3]. This finding led to the rationalization that formulating these drugs as LBDDS could improve bioavailability. LBDDS induce, but to a lower magnitude, the same physiological events seen during meal consumption. Pancreatic and gallbladder secretions initiate lipid digestion and solubilization of digestion products in mixed micelles. Eventual colloidal particles solubilize lipids and coadministered drugs. Based on the foundation, LBDDS are an attractive formulation option for poorly water soluble drugs that experience a positive food effect and we hypothesized that the pharmacokinetics of drugs formulated as LBDDS should not have significant food effects. The general relationship between food effect and BCS Classification states that Class 1 compounds have no food effect, Class 3 have a negative food effect, and Classes 2 and 4 have a positive food effect [Citation44,Citation45]. We reviewed the package inserts to see if there was any mention of food effect or dosing with regard to meals. summarizes the findings. The labels for 21 drug products included language on administration with food. The drugs labeled to be taken with or without food show increased, decreased, or unchanged area under the plasma-time curve (AUC) or Cmax, but the magnitude of change in the pharmacokinetic parameter is not clinically relevant. Of the drugs labeled to be taken with food, eight drugs experience positive food effect and one (lubiprostone) has negative food effect. Lubiprostone is labeled to be taken with food to maintain a lower Cmax. This may be to minimize side effects caused by spikes in serum concentrations.
Table 3. Food effects and pharmacokinetics of approve drugs.
We hypothesize based on our review of approved drugs formulated as LBDDS experience food effect to a lesser degree than drugs with similar solubility properties that are no formulated in lipid-based systems. In support of our hypothesis, we compared the physicochemical properties of solubility (LogP), permeability (effective permeability), and dose for the approved drugs listed in to an analysis of these same parameters (LogP, effective permeability, and dose) in a comparator test set of 15 oral approved drugs consisting of protein kinase inhibitors, BRAF inhibitors, and MEK inhibitors that are not formulated as lipid-based systems. The rationale for selecting the above three parameters (LogP, effective permeability, and dose) is based on previous studies that suggest these three physiochemical properties can be used to accurately categorize the food effect (positive, negative, or neutral) in 80% of a test group of drugs [Citation45]. We wanted to first ensure that the two sets of approved drug were comparable in terms of physicochemical properties that influence food effects (LogP, effective permeability, and dose). To do so, we calculated the median values and performed the two-tailed Mann-Whitney U test with a significance level of 0.05 to compare to difference in median values between the two data sets of approved drugs. The three selected parameters of approved drugs that were not developed as LBDDS, compared to those that were, had the following median values and p values, respectively: LogP 4.02 and 3.85, p values of .13888; effective permeability 6.88 × 10−4 cm/s and 6.92 × 10−4 cm/s, p values of .82588; and median dose 250 mg and 100 mg, p values of .07508. None of the measurements between the two sets were shown to be statistically different (p < .05); therefore, the two data sets are comparable. The median magnitude of food effect on area under the curve for the oral comparator set was +35% (range –31 to +400%) and for the LBDDS set, the median magnitude was +16.5% (range –13 to +100%). However, the difference was not statistically significant with a p value of .63122. Therefore, it cannot be concluded from this exercise that LBDDS blunt the food effect.
Future directions in oral LBDDS delivery
From our analysis, it is evident that lipid formulations are versatile platforms for drugs with a wide range of physicochemical properties. These drugs tend to be lipophilic and permeable thus confirming the widely held belief about ideal candidates to be formulated as lipid formulations. Lipid formulations are commonly used for certain classes of drugs such as retinoids, vitamin D analogs, and protease inhibitors. There are several unexplored areas where lipid formulations may play a role in the future.
Drugs for cardiovascular disease
Currently approved drugs using lipid formulation technology cover many therapeutic categories, but none are used for cardiovascular disease treatment. Cardiovascular drugs are one the most common pharmaceutical categories with more than 100 FDA approved drugs and represent an enticing area for lipid formulations. Rao et al. analyzed the pharmacokinetic profiles of cardiovascular drugs and discovered that over 80% had bioavailability challenges [Citation46]. The primary reasons for these challenges were extensive first pass metabolism and poor water solubility. Because of the low and variable bioavailabilities of some of the best selling drugs, the authors recommended the use of advanced delivery systems to optimize the drugs and presented several case studies demonstrating solubility improvement and enhanced absorption of cardiovascular drugs when formulated into SEDDS. Several cardiovascular drugs experience negative food effects (captopril, moexipril, quinapril, ramipril, losartan, and pravastatin) and may not be the best candidates for lipid formulations.
Oral delivery of peptides
Lipid formulations are used for several high molecular weight molecules including peptides and peptidomimetics. Peptides are becoming increasingly common in clinical practice especially for the treatment of diabetes. Insulin and glucagon like peptide 1 (GLP-1) analogs are typically administered by subcutaneous injections. These peptides have high molecular weights, short half-lives and high clearance. Bioengineering modifications have been used to improve the pharmacokinetics. However, these improved peptides must still be injected.
The use of colloidal carriers offers protection against enzymatic degradation in the gastrointestinal tract [Citation47]. Microemulsions are more likely than polymeric carriers to be developed into an approved oral drug product, indicated by the number of approvals using each approach. For example, peptides are hydrophilic drugs that can be classified as BCS Class III compounds. Permeation and stability in the GI tract are the primary challenges to the oral absorption of peptides. As a result of their hydrophilic nature, peptides can reside in the aqueous phase of water-in-oil (w/o) microemulsions. The oil phase can minimize exposure to water and protect against degradation. Surfactants, co-surfactants, and medium chain diacylglycerols in the formulation may improve membrane permeability and therefore enhance peptide drug bioavailability. Furthermore, there is potential to reduce peptide degradation in the acidic stomach and the enzymatically active regions of the gastrointestinal tract given the protection afforded by a w/o microemulsion [Citation48–50].
Oral anticancer drugs
Protein kinase inhibitors have led the way in the development of oral anticancer agents. Compared with traditional chemotherapeutic agents, these newer drugs are targeted, have better side effect profiles, and are administered orally and chronically. The majority of these drugs have poor water solubility and significant food effects. More specifically, the majority of these agents have a positive food effect and are labeled to be taken on an empty stomach, 2 h before or 1 h after a meal [Citation51,Citation52]. The increase in AUC and Cmax due to food can result in supratherapeutic and toxic concentrations of the anticancer drugs, a number of which carry FDA black box warnings for potentially life-threatening risks associated with the drug [Citation53]. One of the most common and serious side effects would be QTc prolongation, potentially creating dangerous cardiac arrhythmias. Considering that patients with chronic disease typically take multiple medications for multiple conditions, they may not be able to adhere to a strict guideline of taking a medication on an empty stomach. Lipid formulations are known to mimic, to a certain extent, the high fat content of a meal and increase solubilization of the drug molecule [Citation54]. Therefore, lipid formulations of anticancer agents have the potential to improve the absorption and minimize the food effect of anticancer agents thereby eliminating safety concerns arising from lack of patient adherence.
Amphotericin B
Amphotericin B is a polyene macrolide antibiotic used in the treatment of serious systemic fungal infections, acting by binding to fungal cell wall sterols. The drug has amphoteric features as well as hydrophobic and hydrophilic portions of this relatively large, small-molecule drug (MW= 924). It has pKa’s of 2, 5.5 and 10.0. At 28 °C, its water solubility is only 0.75 mg/mL and it is also practically insoluble in ethanol [Citation55].
To understand the goals and limitations of developing the highly desirable oral formulation of Amphotericin B, it is important to understand the history and biophysical behavior of LBDDS intravenous formulations. The deoxycholate formulation (Fungizone® injection) was developed in the 1970 s to address the solubility problems; however, its use is limited by infusion-related reactions and dose-related nephrotoxicity. To reduce renal toxicity and increase the therapeutic efficacy of Amphotericin B, intravenous lipid formulations of Amphotericin B including Ablecet (1995), Amphotec (1996), and Ambisome (1997) were approved [Citation56]. Abelcet (Amphotericin B lipid complex injection) is comprised of 5 mg of Amphotericin B per dose with the lipid excipients L-α-dimyristoylphosphatidylcholine (DMPC, 3.4 mg) and L-α-dimyristoylphosphatidylglycerol (DMPG, 1.5 mg) [Citation57]. The complex forms a stable ribbon-like structure with Amphotericin B with a heterogeneous 2–5 μm diameter size range [Citation58,Citation59], which is hypothesized to be responsible for its altered biodistribution and toxicity profile compared to the desoxycholate formation. An alternative LBDDS was Amphotec/Amphocil, which is comprised of disc-like structures containing cholesterol sulfate that are significantly smaller, at around 120 nm [Citation60]. Ambisome is comprised of unilamellar phospholipid liposomes <100 nm in mean diameter, comprised of hydrogenated soy phosphatidylcholine and distearoyl phosphatidylglycerol. The size and composition of the structures is important because it determines not only stability in circulation but also organ biodistribution, with a significant portion of the dose going to organs of the reticuloendothelial system.
While these lipid-formulations have been very successful, the major limitations of their use are that they must be administered intravenously and they are very expensive. Hyperkalemia and other electrolyte disturbances can be a problem with liposomal Amphotericin B [Citation61]. The oral bioavailability of Amphotericin B is <5% and for the deoxycholate form is only 2% [Citation62]. Barriers to oral absorption are considerable due to its lack of both solubility and lipophilicity. However, LBDDS are ideal for Amphotericin B in order to achieve not only improved oral absorption but also the desired biodistribution as discussed above, by accessing the lymphatic transport pathways utilized by dietary lipids [Citation63–67]. When suspended in Peceol® (glyceryl monooleate), amphotericin B (AmpB)’s bioavailability is significantly improved [Citation68–70]. AmpB in a Peceol/Gelucire 44/14 (lauroyl macrogol-32 glycerides)-based formulation (iCo -010) has demonstrated efficacy in several animal models of systemic fungal infections [Citation71] as well as visceral leishmaniasis [Citation72,Citation73]. Organ distribution of amphotericin B is shifted away from the kidney, the site of dose-limiting toxicity, with a corresponding increased splenic and hepatic uptake when the drug is administered as LBDDS [Citation67].
An alternative LBDDS is the multi-layered, spiral-shaped cochleate formulation [Citation74]. Precipitates of phosphatidylserine and calcium incorporating amphotericin B were prepared by Sesana et al. [Citation75] and demonstrated to have in vitro activity against Leishmania chagasi. However, further development of amphotericin B lipid cochleates has been limited, perhaps in part due to the low bioavailability of the cochleate formulation [Citation76]. There are additional LBDDS in preclinical testing that have promising improvements in bioavailability as well, indicated in . It remains to be seen which oral formulation is eventually developed and accepted. This will depend not only differential efficacy and toxicity in humans, but also the ease of manufacturing scale-up and market competition.
Table 4. Oral amphotericin formulations in development.
Conclusion
LBDDS are the most studied bioavailability enhancing technology and have been utilized in dozens of FDA approved drugs. Our primary focus was to analyze approved drug molecules that benefited from being formulated in LBDDS. We examined the physicochemical characteristics of these drugs, and we classified them according to different classification systems (BCS, DCS, and LFCS) to see if there were any correlations between physicochemical characteristics and classification systems or between classification systems. Our analysis of the properties of FDA approved drugs showed a diverse set of drug molecules including a range of molecular weights, solubilities, PSA, and chemical structures. As expected, the majority of these molecules have poor solubility and good permeability. We introduced a new physicochemical parameter of octanol solubility, as a surrogate for drug solubility in lipid systems/solvent capacity, and demonstrated its potential value for guiding lipid formulation. Specifically, logarithm of the octanol solubility (Log[Oct]) has a linear relationship with the predicted LogP not related to simply poor water solubility. A Log[Oct] value >2.5 may be a more useful predictor of suitability in LBDDS than LogP alone. Although the analysis revealed some clear emerging trends with respect to choosing the best lipid formulation type based on DCS class, e.g. DCS class I molecules are typically formulated as Type I lipid formulations, the dataset would benefit from expansion in order to make a firm recommendation. It is hoped that future studies will expand this dataset with an integrated assessment of the various physicochemical properties to give the best guidance for new oral drug formulation design.
Table S1
Download PDF (42.4 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
RS, JB, and VP are employees of Catalent Pharma Solutions.
References
- Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129:1–10.
- Crew M. Bioavailability enhancement - analysis of the Historical Use of Solubilization Technologies. Drug Development & Delivery. 2014. Available from: http://www.drug-dev.com/Main/Back-Issues/BIOAVAILABILITY-ENHANCEMENT-Analysis-of-the-Histor-657.aspx#sthash.xOZ5heIQ.dpuf
- Feeney OM, Crum MF, McEvoy CL, et al. 50years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv Drug Deliv Rev. 2016;101:167–194.
- Strickley RG. Currently marketed oral lipid-based dosage forms: drug products and excipients. In: Hauss D, editor. Oral lipid based formulations. 1st ed. New York: Informa Healthcare; 2007.
- Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672.
- Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21:201–230.
- Agrawal SGT, Tripathi DK, Alexander A. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant based self micro emulsifying drug delivery system: a novel approach. Am J Drug Discov Dev. 2012;2:143–183.
- Porter CJH. Oral lipid-based formulations: Using pre-clinical data to dictate formulation strategies for poorly water soluble drugs. In: Hauss D, editor. Oral lipid-based formulations: enhancing the bioavailability of poorly water-soluble drugs. New York Informa Healthcare; 2007. p. 185–205.
- Gullapalli RP. Soft gelatin capsules (softgels). J Pharm Sci. 2010;99:4107–4148.
- Jones WJ, 3rd, Francis JJ. Softgels: consumer perceptions and market impact relative to other oral dosage forms. Adv Ther. 2000;17:213–221.
- Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287.
- Griffin B. Advances in lipid-based formulations: overcoming the challenge of low bioavailability for poorly water soluble drug compounds. Am Pharm Rev. March 2012. Available from: http://www.americanpharmaceuticalreview.com/Featured-Articles/39299-Advances-in-Lipid-based-Formulations-Overcoming-the-Challenge-of-Low-Bioavailability-for-Poorly-Water-Soluble-Drug-Compounds/
- MacGregor KJ, Embleton JK, Lacy JE, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25:33–46.
- Kahan BD, Dunn J, Fitts C, et al. Reduced inter- and intrasubject variability in cyclosporine pharmacokinetics in renal transplant recipients treated with a microemulsion formulation in conjunction with fasting, low-fat meals, or high-fat meals. Transplantation 1995;59:505–511.
- Mendez R, Abboud H, Burdick J, et al. Reduced intrapatient variability of cyclosporine pharmacokinetics in renal transplant recipients switched from oral sandimmune to neoral. Clin. Ther.. 1999;21:160–171.
- Katneni K, Charman SA, Porter CJ. Impact of cremophor-EL and polysorbate-80 on digoxin permeability across rat jejunum: delineation of thermodynamic and transporter related events using the reciprocal permeability approach. J Pharm Sci. 2007;96:280–293.
- Wacher VJ, Silverman JA, Zhang Y, et al. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87:1322–1330.
- Kim AE, Dintaman JM, Waddell DS, et al. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J Pharmacol Exp Ther. 1998;286:1439–1445.
- Schmitt C, Kaeser B, Riek M, et al. Effect of saquinavir/ritonavir on P-glycoprotein activity in healthy volunteers using digoxin as a probe. CP. 2010;48:192–199.
- Perloff MD, Von Moltke LL, Marchand JE, et al. Ritonavir induces P-glycoprotein expression, multidrug resistance-associated protein (MRP1) expression, and drug transporter-mediated activity in a human intestinal cell line. J Pharm Sci. 2001;90:1829–1837.
- Chen XQ, Gudmundsson OS, Hageman MJ. Application of lipid-based formulations in drug discovery. J Med Chem. 2012;55:7945–7956.
- Gao P. MW. Case studies: development of self-emulsifying formulations for improving the oral bioavailability of poorly soluble, lipophilic drugs. In: DJ H, editor. Oral lipid-based formulations enhancing the bioavailability of poorly water soluble drugs. 1st ed. New York: Informa Healthcare; 2007.
- Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60:625–637.
- Backman TW, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011;39:W486–W491.
- Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/in vivo case studies. J Pharm Sci. 2007;96:235–248.
- Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14:10–18.
- Akhtar N, Ahad A, Khar RK, et al. The emerging role of P-glycoprotein inhibitors in drug delivery: a patent review. Expert Opin Ther Pat. 2011;21:561–576.
- Kayser O, Olbrich C, Yardley V, et al. Formulation of amphotericin B as nanosuspension for oral administration. Int J Pharm. 2003;254:73–75.
- Lindmark T, Kimura Y, Artursson P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J Pharmacol Exp Ther. 1998;284:362–369.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623.
- Hou TJ, Zhang W, Xia K, et al. ADME evaluation in drug discovery. 5. Correlation of Caco-2 permeation with simple molecular properties. J Chem Inf Comput Sci. 2004;44:1585–1600.
- Flaten GE, Luthman K, Vasskog T, et al. Drug permeability across a phospholipid vesicle-based barrier 4. The effect of tensides, co-solvents and pH changes on barrier integrity and on drug permeability. Eur J Pharm Sci. 2008;34:173–180.
- Cornaire G, Woodley JF, Saivin S, et al. Effect of polyoxyl 35 castor oil and Polysorbate 80 on the intestinal absorption of digoxin in vitro. Arzneimittelforschung 2000;50:576–579.
- Mullertz A, Ogbonna A, Ren S, et al. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62:1622–1636.
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890.
- Adkins JC, Faulds D. Amprenavir. Drugs 1998;55:837–842. discussion 43-4.
- Amidon GL, Lennernas H, Shah VP, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420.
- Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci. 2010;99:4940–4954.
- Dahan A, Hoffman A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67:96–105.
- Porter CJ, Kaukonen AM, Taillardat-Bertschinger A, et al. Use of in vitro lipid digestion data to explain the in vivo performance of triglyceride-based oral lipid formulations of poorly water-soluble drugs: studies with halofantrine. J Pharm Sci. 2004;93:1110–1121.
- Lipinski CA, Lombardo F, Dominy BW, et al. approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26.
- Bergstrom CA, Charman WN, Porter CJ. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv Drug Deliv Rev. 2016;101:6–21.
- Leeson PD. Molecular inflation, attrition and the rule of five. Adv Drug Deliv Rev. 2016;101:22–33.
- Fleisher D, Li C, Zhou Y, et al. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clin Pharmacokinet. 1999;36:233–254.
- Gu CH, Li H, Levons J, et al. Predicting effect of food on extent of drug absorption based on physicochemical properties. Pharm Res. 2007;24:1118–1130.
- Rao S, Tan A, Thomas N, et al. Perspective and potential of oral lipid-based delivery to optimize pharmacological therapies against cardiovascular diseases. J Control Release. 2014;193:174–187.
- Martins S, Sarmento B, Ferreira DC, et al. Lipid-based colloidal carriers for peptide and protein delivery – liposomes versus lipid nanoparticles. Int J Nanomed. 2007;2:595–607.
- Hintzen F, Perera G, Hauptstein S, et al. In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int J Pharm. 2014;472:20–26.
- Liu D, Kobayashi T, Russo S, et al. In vitro and in vivo evaluation of a water-in-oil microemulsion system for enhanced peptide intestinal delivery. AAPS J. 2013;15:288–298.
- Guo L, Ma E, Zhao H, et al. Preliminary evaluation of a novel oral delivery system for rhPTH1-34: in vitro and in vivo. Int J Pharm. 2011;420:172–179.
- Ratain MJ. Flushing oral oncology drugs down the toilet. J Clin Oncol. 2011;29:3958–3959.
- Kang SP, Ratain MJ. Inconsistent labeling of food effect for oral agents across therapeutic areas: differences between oncology and non-oncology products. Clin Cancer Res. 2010;16:4446–4451.
- Food and Drug Administration. Drug Watch. [cited 2016 Nov 4]. Available at: https://www.drugwatch.com/2012/01/18/fda-black-box-warnings/
- Cerpnjak K, Zvonar A, Gasperlin M, et al. Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharmaceutica (Zagreb, Croatia) 2013;63:427–445.
- Yalkowsky SH. Handbook of Aqueous Solubility Data: An Extensive Compilation of Aqueous Solubility Data for Organic Compounds Extracted from the AQUASOL dATAbASE. Boca Raton, Florida: CRC Press LLC; 2003. p. 1263.
- Food and Drug Administration. Drugs. [cited 2016 Nov 4]. Available from: http://www.fda.gov/Drugs/default.htm
- Abelcet® [package insert]. Gaithersburg, MD: Sigma-Tau Pharmaceuticals Inc; 2013.
- Janoff AS, Boni LT, Popescu MC, et al. Unusual lipid structures selectively reduce the toxicity of amphotericin B. Proc Natl Acad Sci USA. 1988;85:6122–6126.
- Mistro S, Maciel Ide M, de Menezes RG, et al. Does lipid emulsion reduce amphotericin B nephrotoxicity? A systematic review and meta-analysis. Clin Infect Dis. 2012;54:1774–1777.
- Guo LS. Amphotericin B colloidal dispersion: an improved antifungal therapy. Adv Drug Deliv Rev. 2001;47:149–163.
- Johansen HK, Gotzsche PC. Amphotericin B lipid soluble formulations versus amphotericin B in cancer patients with neutropenia. Cochrane Database Syst Rev 2014;(9):CD000969.
- Chen Y-C, Su C-Y, Jhan H-J, et al. Physical characterization and in vivo pharmacokinetic study of self-assembling amphotericin B-loaded lecithin-based mixed polymeric micelles. Int J Nanomed. 2015;10:7265–7274.
- Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–248.
- Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60:702–716.
- Yanez JA, Wang SW, Knemeyer IW, et al. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011;63:923–942.
- Zgair A, Wong JC, Lee JB, et al. Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. Am J Trans Res. 2016;8:3448–3459.
- Sachs-Barrable K, Lee SD, Wasan EK, et al. Enhancing drug absorption using lipids: a case study presenting the development and pharmacological evaluation of a novel lipid-based oral amphotericin B formulation for the treatment of systemic fungal infections. Adv Drug Deliv Rev. 2008;60:692–701.
- Risovic V, Boyd M, Choo E, et al. Effects of lipid-based oral formulations on plasma and tissue amphotericin B concentrations and renal toxicity in male rats. Antimicrob Agents Chemother. 2003;47:3339–3342.
- Gershkovich P, Wasan EK, Lin M, et al. Pharmacokinetics and biodistribution of amphotericin B in rats following oral administration in a novel lipid-based formulation. J Antimicrob Chemother. 2009;64:101–108.
- Sivak O, Gershkovich P, Lin M, et al. Tropically stable novel oral lipid formulation of amphotericin B (iCo-010): biodistribution and toxicity in a mouse model. Lipids Health Dis. 2011;10:135. Epub 2011/08/10.
- Risovic V, Rosland M, Sivak O, et al. Assessing the antifungal activity of a new oral lipid-based amphotericin B formulation following administration to rats infected with Aspergillus fumigatus. Drug Dev Ind Pharm. 2007;33:703–707.
- Gershkovich P, Wasan EK, Sivak O, et al. Visceral leishmaniasis affects liver and spleen concentrations of amphotericin B following administration to mice. J Antimicrob Chemother. 2010;65:535–537.
- Wasan EK, Gershkovich P, Zhao J, et al. A novel tropically stable oral amphotericin B formulation (iCo-010) exhibits efficacy against visceral Leishmaniasis in a murine model. PLoS Negl Trop Dis. 2010;4:e913.
- Perlin DS. Amphotericin B cochleates: a vehicle for oral delivery. Curr Opin Investig Drugs. 2004;5:198–201.
- Sesana AM, Monti-Rocha R, Vinhas SA, et al. In vitro activity of amphotericin B cochleates against Leishmania chagasi. Mem Inst Oswaldo Cruz. 2011;106:251–253.
- Lemke A, Kiderlen AF, Kayser O. Amphotericin B. Appl Microbiol Biotechnol. 2005;68:151–162.
- Gupta PK, Jaiswal AK, Asthana S, et al. Synergistic enhancement of parasiticidal activity of amphotericin B using copaiba oil in nanoemulsified carrier for oral delivery: an approach for non-toxic chemotherapy. Br J Pharmacol. 2015;172:3596–3610.
- Yang Z, Chen M, Yang M, et al. Evaluating the potential of cubosomal nanoparticles for oral delivery of amphotericin B in treating fungal infection. Int J Nanomed. 2014;9:327–336.
- Yang Z, Tan Y, Chen M, et al. Development of amphotericin B-loaded cubosomes through the SolEmuls technology for enhancing the oral bioavailability. AAPS PharmSciTech. 2012;13:1483–1491.
- Delmas G, Park S, Chen ZW, et al. Efficacy of orally delivered cochleates containing amphotericin B in a murine model of Aspergillosis. Antimicrob Agents Chemother. 2002;46:2704–2707.