
Abstract
In recent years, the incidence and mortality rates of lymphoma have gradually increased worldwide. Tumorigenesis and drug resistance are closely related to intracellular inflammatory pathways in lymphoma. Therefore, understanding the biological role of inflammatory pathways and their abnormal activation in relation to the development of lymphoma and their selective modulation may open new avenues for targeted therapy of lymphoma. The biological functions of inflammatory pathways are extensive, and they are central hubs for regulating inflammatory responses, immune responses, and the tumour immune microenvironment. However, limited studies have investigated the role of inflammatory pathways in lymphoma development. This review summarizes the relationship between abnormal activation of common inflammatory pathways and lymphoma development to identify precise and efficient targeted therapeutic options for patients with advanced, drug-resistant lymphoma.
KEY MESSAGES
Inflammatory pathways directly or indirectly regulate the TME and are closely related to the development of lymphoma.
This review was conducted to elucidate the connection between inflammatory pathways and the tumorigenesis and drug resistance of several common lymphomas.
Overall, targeting abnormally activated molecules upstream and downstream of lymphoma inflammatory pathways in the future is expected to be a new target for lymphoma treatment.
Introduction
Lymphoma is a malignancy originating from the lymphopoietic system and is classified as Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL). Approximately 10%–15% of lymphoma cases account for HL, whereas 85%–90% of cases account for NHL [Citation1]. The morbidity rate of patients with NHL has increased in recent years [Citation2–4]. The mortality rate of lymphoma is increasing by 4.5% annually, increasing with the age of patients [Citation5]. Lymphomas are malignant tumours with diverse patterns of clinical behaviour that are clonally proliferated by lymphocytes, with biological heterogeneity leading to marked differences in the epidemiology, pathological staging, and optimal treatment of lymphomas [Citation6, Citation7]. Consequently, lymphoma is a major public health concern worldwide. Genetic mutations and interactions between tumour and immune cells in the tumour microenvironment (TME) partly lead to the activation of many inflammatory pathways and regulate the upstream and downstream targets of the pathways in a complex manner in lymphoma. The TME consists of tumour-associated cells and stroma, including different types of immune cells, cancer-associated fibroblasts (CAFs), tumour-associated macrophages (TAMs), various cytokines, and extracellular matrix [Citation8]. The TME is complex owing to interactions between tumour cells and plays an essential role in tumour growth, invasion, and metastasis [Citation8, Citation9]. Inflammatory pathways directly or indirectly regulate the TME and are closely related to the development of lymphoma [Citation10–12]. Therefore, their role in the pathophysiology of lymphoma has received substantial attention. The signalling pathways known to be involved in the development of lymphoma include the JAK/STAT, NF-κB, PI3K/AKT/mTOR, Toll-like receptor (TLR), B-cell receptor (BCR), and T-cell receptor signalling pathways. This review focuses on the mechanisms of the JAK/STAT, PI3K/AKT/mTOR, and NF/κB signalling pathways in HL and several common NHL to provide a reference for developing effective treatment strategies in the future.
1. JAK/STATs signaling pathway
In the JAK/STAT signalling pathway, receptors bind to various intracellular cytokines to initiate the transcription of related genes, thereby regulating cell proliferation, inflammation, differentiation, and apoptosis [Citation13]. It primarily comprises receptor tyrosine kinases (RTKs), Janus tyrosine kinases (JAKs), and signal transducer and activator of transcription proteins (STATs) [Citation14]. When cytokines bind with their receptors, JAKs are triggered and transmit regulatory signals. STATs are responsible for cytokine signalling in cells and are delivered to the nucleus to initiate transcription. STATs contain six structural domains and different structural domains modulate diverse features of STATs [Citation15–17]. The conserved N-terminal structural domain drives the formation of STAT dimers, allowing them to bind to transcription factors [Citation18]. The superhelix domain is associated with regulatory proteins and is involved in controlling the entry and exit of material from the nucleus [Citation19, Citation20]. The DNA-binding domain binds to specific target gene sequences, and the SH2 structural domain is mainly responsible for recognizing the phosphotyrosine motifs of cytokine receptors and dimerising STATs with activated JAKs [Citation21, Citation22]. The C-terminal trans-activation domain contains phosphorylation sites, which activate STATs and promote the transcription of target genes.
The JAK/STAT signalling pathway promotes cytokine-mediated gene transcription in a simple and efficient way. JAKs non-covalently link to cytokine receptors, which coordinate receptor tyrosine phosphorylation and recruit STATs [Citation14]. Phosphorylated STATs form homodimers or heterodimers with other STAT members harbouring conserved SH2 structural domains [Citation23], which are subsequently transported across the nuclear membrane to the nucleus and bind to specific regulatory DNA sequences to regulate the expression of specific genes, including those related to cell survival, proliferation, angiogenesis, and invasion [Citation24–26].(Fig1A)The JAK/STAT signalling pathway mediates immune responses and is involved in the maintenance of environmental homeostasis in the lymphatic system [Citation23]. It functions to maintain the balance between Th1 and Th2 cells during the immune response and affects the development of regulatory T cells(Tregs)and the function of memory CD8+ T cells [Citation27]. The JAK/STAT signalling pathway is widely found in different types of tissues and cells and is considered one of the central communication nodes for cellular function [Citation14]. Its over-activation is reported to be closely related to the growth, metastasis, and invasion of various tumours. For example, in STAT6VT transgenic mice, with a mutation in the SH2 domain (STAT6V547A/T548A), sustained activation of STAT6 promotes Th2 cell differentiation, increases IgG1 and IgE levels in serum, and decreases the abundance of T cells and increases the abundance of B cells in peripheral blood, leading to immune system disorder and the development of spontaneous lymphoma [Citation28].
Recent studies have demonstrated that the JAK/STAT signalling pathway is commonly aberrantly activated in various lymphomas, including HL, MCL, DLBCL, primary mediastinal large B-cell lymphoma (PMBCL), cutaneous T-cell lymphoma (CTCL), BL, and NK/T-cell lymphoma (NKTL). In a case study on HL and PMBCL, approximately 30% of patients with HL and PMBCL had abnormally high JAK2 expression [Citation29]. These two lymphomas have high morbidity in adolescents and are similar in terms of clinical presentation, pathological and histological changes, and pathogenesis [Citation30–32]. Tumour cells are surrounded by a large number of infiltrated inflammatory cells, suggesting that tumour cells may escape immune surveillance. Green et al. (2010) reported that the abovementioned phenomenon is attributed to the abnormal amplification of the 9p24.1 gene in tumour cell chromosomes, which leads to the increased expression of programmed cell death ligand 1 (PD-L1) and JAK2 at 9p24.1 [Citation29]. The continuous activation of the JAK/STAT signalling pathway further promotes the increase in PD-L1 expression, and the highly expressed PD-L1 binds to the corresponding receptors on the surface of T cells, which causes T cells to ‘over-deplete’ and lose their function, resulting in the escape of tumour cells from immune surveillance [Citation29, Citation33]. (Fig1B) Cutaneous T-cell lymphoma (CTCL) is a heterogeneous group of malignant lymphoid tissue proliferative diseases mediated by tumorigenic T cells [Citation34–36]. Tumour cells secrete IL-13, IL-22, and oncostatin M, activate the STAT3 signalling pathway, and down-regulate the expression of filaggrin and filaggrin-2 in human keratinocytes [Citation37]. The primary function of the epidermis is to act as a barrier to the outside world, and it is extremely important to ensure the barrier function and integrity of the stratum corneum [Citation38]. Deficiency of filaggrin leads to reduced barrier function, bacterial colonization, dysregulated microbial ecology and increased susceptibility to infection [Citation39, Citation40]. Approximately 25% of patients with Natural killer/T-cell lymphoma (NKTCL) have the A572V mutation in the JH2 structural domain of JAK3. Compared with wild-type JAK3, mutated JAK3 protein has an extended half-life, resulting in increased JAK3 kinase activity and aberrant activation of downstream STAT5 [Citation41]. Moreover, treatment of JAK 3 mutant and wild-type NKTCL cell lines with the novel pan-JAK inhibitor CP-690550 [Citation42] revealed a decrease in phosphorylated STAT 5, a decreased cell viability, and an enhanced apoptosis [Citation41]. Thus, targeting the dysregulated JAK/STAT pathway may be a promising therapeutic approach for NKTCL patients. Meanwhile, Rivera-Munoz et al. (2018) demonstrate that activated JAK3 leads to a progressive and dose-dependent expansion of CD8 + T cells in the periphery before colonization of the bone marrow by a JAK3A572Vknockin mouse model. This model presents several features of the human leukemic form of cutaneous T-cell lymphoma (L-CTCL), including skin involvements [Citation43]. It was proved that aberrant activation of JAK3 due to A572 mutation was also existed in CTCL. (Fig1B) Cornejo et al. (2009) demonstrated that sustained activation of the JAK/STAT signalling pathway increased the number of CD8 + TCRαβ + CD44 + CD122 + Ly-6C + T cells in a mouse model of bone marrow transplantation. Compared with wild-type cells, these aberrant T cells exhibit increased proliferative capacity; enhanced anti-apoptotic capacity; and increased expression of c-myc, Bcl-2, interferon-gamma, and tumour necrosis factor-alpha, resulting in life-threatening lymphoid tissue proliferation [Citation44].
STAT3, STAT5, and STAT6 are activated and highly expressed in HL, indicating that the JAK/STAT signalling pathway is a crucial driver of HL pathogenesis. At the same time, Bcl-xL is an anti-apoptotic gene whose expression is induced by the binding of STAT5 DNA, and activation of JAK2/STAT5 pathway can regulate apoptosis and survival through Bcl-Xl [Citation45]. STAT6 and SOCS1 are the two most frequent mutation targets (32% and 59%, respectively). Missense mutations in the DNA-binding domain of STAT6, including hotspot regions such as N417, D419, and N421, are detected in 32% of cases [Citation46, Citation47]. (Fig1B) These mutations considerably overlap with those in PMBCL, relapsed or refractory diffuse large B-cell lymphoma (rrDLBCL) with GCB type and follicular lymphoma (FL) [Citation28, Citation48–50]. In addition, tumour cells highly express and secrete several cytokines that contribute to increased STAT phosphorylation, which in turn results in sustained activity of the JAK/STAT signalling pathway and accelerates tumour progression [Citation51]. The same mechanism has been observed in MCL. MCL cells have high expression of the membrane protein GP130 and autocrine production of high concentrations of GP80 and IL-6, leading to continuous activation of the JAK/STAT signalling pathway [Citation52]. With improved recognition of the biology of DLBCL and the application of R-CHOP treatment protocols, the prognosis of DLBCL has significantly improved; however, that of rrDLBCL remains poor, and only 45% patients achieve long-term remission with salvage chemotherapy and autologous stem cell transplantation (SCT) [Citation53]. Therefore, investigating the mechanisms underlying the development of drug resistance in rrDLBCL is necessary. Morin et al. (2015) found high-frequency mutations in STAT6 in 38 patients with rrDLBCL. These mutations may be responsible for treatment resistance in rrDLBCL [Citation47]. Koo et al. (2012) reported that 23 of 65 (35.4%) patients with NK/TCL had a mutation in JAK3, which caused persistent activation of the JAK/STAT signalling pathway independent of stimulation by external signalling molecules and promoted cell proliferation [Citation41]. This finding suggests that the development of NK/TCL is related to the abnormal activation of the JAK/STAT signalling pathway. A study reported that the most frequently mutated gene was STAT3 (37%) in patients with plasmablastic lymphoma (PBL) [54].
Currently, based on the understanding of the role of the JAK/STAT signalling pathway in the development of several common lymphomas, the search for immune checkpoints and the development of immunosuppressive agents to improve the prognosis of patients has become a top priority. The PD-1 inhibitors nivolumab and pembrolizumab have both demonstrated high remission rates in patients with relapsed/refractory HL and have been widely used in the clinic [Citation55]. Lestaurtinib, as an oral multikinase inhibitor, has been shown to inhibit JAK2 in myeloproliferative disorders. Diaz et al. (2011) analyzed the treatment of five refractory HL cell lines with Lestaurtinib and found that cell growth inhibition and apoptosis were dose-dependent at 48h [Citation56]. This finding provides a molecular theoretical basis for the use of JAK2 inhibitors, especially Lestaurtinib, in HL patients.
In conclusion, the JAK/STAT signalling pathway plays an important role in the development of lymphoma. () Blocking the upstream molecules that cause abnormal activation of the JAK/STAT signalling pathway may represent a novel strategy for the treatment of lymphoma. In addition, JAKs can inhibit the PI3K/AKT signalling pathway. Phosphorylated JAKs activate PI3K, which in turn triggers its downstream cascades.
Table 1. Mutation or overexpression of JAK/STAT at different lymphoma diseases.
2. PI3K/AKT/mTOR signalling pathway
The PI3K–AKT–mTOR signalling pathway primarily comprises type I phosphatidylinositol 3-kinase (PI3K) and its downstream molecules serine/threonine protein kinase B (PKB/Akt) and mammalian target of rapamycin (mTOR) [Citation57, Citation58]. Members of the PI3K family are proto-oncogenes; in particular, type I PI3K is a heterodimer that is widely mutated in tumour cells. Type I PI3K is composed of the regulatory subunit P85 and the catalytic subunit P110 [Citation59]. It can be divided into two subtypes, IA and IB, which are activated in two major ways: (1) interaction of PI3K with growth factor receptors or ligand proteins that have tyrosine residues and are phosphorylated (2) direct binding of p110 through Ras. Activated PI3K activates AKT by phosphorylating its substrate phosphatidylinositol 3,4-biphosphate (PIP2) to phosphatidylinositol 3,4,5-biphosphate (PIP3) [Citation60]. In addition, it activates the mTOR protein through phosphorylation. mTOR includes two polymolecular complexes, namely, mTORC1 and mTORC2, which regulate protein synthesis and cell growth.
The PI3K/AKT/mTOR signalling pathway is involved in protein synthesis and is responsible for cell growth, differentiation, metabolism, survival, and proliferation [Citation61]. Aberrant activation of the PI3K–AKT–mTOR signalling pathway is an essential carcinogenic signal [Citation62]. Activation of the pathway enhances the ability of tumour cells to proliferate, grow, and participate in angiogenesis. And this will lead to tumour cell escape and tumour invasion and migration [Citation63]. In the PI3K–AKT–mTOR pathway, the components are tightly connected and are essential for regulating the cell cycle and interacting with several other pathways, such as the hypoxia-inducible factor (HIF), mitogen-activated protein kinase (MAPK), Notch, Wnt, Jun, and Ras pathways [Citation64] ().
Figure 1. (A) Activation and regulation of the canonical JAK/STAT signalling pathway: cytokine binding to its receptor recruits and activates JAKs coupled to the receptor, which phosphorylates specific tyrosines on the bound receptor and creates docking sites for proteins with the SH2 structural domain (including STATs). Subsequently, JAKs mediate the phosphorylation of STATs to form homodimers/heterodimers into the nucleus, which bind to the promoters of the corresponding target genes to activate the transcription and expression of the relevant genes. (B) Abnormal activation of the JAK/STAT pathway in lymphoma: 1. In HL and PMBCL, the abnormal amplification of the 9p24.1 gene in the chromosome of tumour cells leads to an increase in the expression of PD-L1 and JAK2 at this position, and the continuous activation of the JAK/STAT signalling pathway further promotes the increase in the expression of PD-L1, and the high-expression of PD-L1 binds to PD-1 on the surface of the T cells, causing the T cells to ‘over-deplete’ and loss function of T cells, leading to the escape of tumour cells from immune surveillance. 2. In CTCL, JAK3 mutations lead to an increase in activity and an abnormal activation of the downstream STAT5, while a variety of cytokines secreted by the tumour cells also lead to the upregulation of the constitutive activation of the STAT5, leading to the survival and development of malignant tumour cells.3. In HL, STAT3, STAT5, STAT6 can be abnormally activated and expressed at high levels, accelerating tumour development.
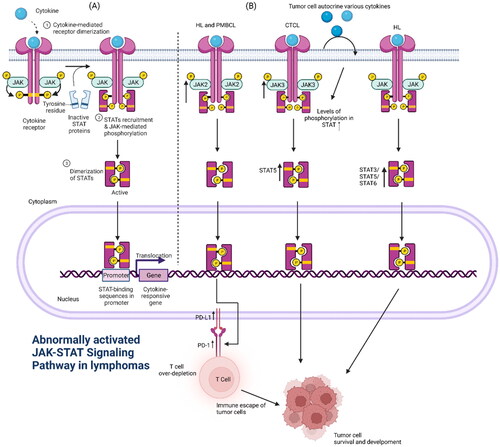
Figure 2. Type I PI3K is composed of the regulatory subunit P85 and the catalytic subunit P110. It can be divided into two subtypes, IA and IB, which are activated in two major ways: (1) interaction of PI3K with growth factor receptors or ligand proteins that have tyrosine residues and are phosphorylated (2) direct binding of p110 through Ras. Activated PI3K activates AKT by phosphorylating its substrate PIP2 to PIP3. In addition, it activates the mTOR protein through phosphorylation. mTOR includes two polymolecular complexes, namely, mTORC1 and mTORC2, which regulate protein synthesis and cell growth. Abnormal activation of the PI3K/AKT/mTOR pathway in lymphoma: 1. Mutations in the PIK3CA gene encoding p110α in patients with DLBCL and increased copy number of PIK3CA in patients with MCL lead to aberrant activation of the PI3K/AKT/mTOR pathway.2 In some patients with lymphoma, the phosphorylation of AKT is markedly increased, which leads to the inhibition of the function of the activated AKT on the cell-cycle inhibitors P21 and P27, resulting in uncontrolled tumour cells growth.3 High expression of AKT directly phosphorylates downstream FOX03a, which is associated with poor prognosis in patients with MCL.3 PTEN function, which is opposite to PIK3, inhibits AKT function, and inhibits downstream FOX03a, which is associated with poor prognosis in patients with MCL. The high expression of AKT directly phosphorylates downstream FOX03a, which is significantly associated with poor prognosis in MCL patients.3. PTEN functions in the opposite way to PIK3, inhibiting AKT activation and blocking signalling, and the lack of PTEN in GCB-type DLBCL patients leads to aberrant activation of the PI3K/AKT/mTOR signalling pathway.
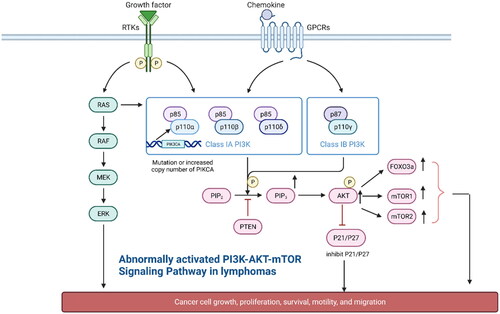
Aberrant activation of the PI3K–AKT–mTOR pathway in lymphoma can be attributed to the following reasons: (1) Mutation or gene amplification of PI3K subtypes can lead to elevated pathway activity. PIK3CA, the gene encoding p110a, is mutated in 10% of patients with DLBCL [Citation65]. It also indicates that targeted small molecule antagonists of the PIK3CA protein may become one of the effective measures for the treatment of DLBCL. Psyrri et al. (2009) reported that the copy number of PIKCA was increased in most patients with MCL, which led to pathway activation and contributed to the progression of MCL [Citation66]. This also raises the possibility that PIK3CA could serve as a molecular target for MCL. (2) AKT phosphorylation indicates activation of the PI3K pathway, which is a common occurrence in lymphoma [Citation67]. Activated AKT inhibits the function of the cell cycle inhibitors P21 and P27, resulting in uncontrolled tumour cell growth [Citation68, Citation69]. Georgakis et al. (2006) detected AKT phosphorylation in 63% of biopsy samples from patients with HL [Citation70] and 52% of patients with DLBCL [Citation71]. Xiaowei et al. (2022) reported that the PI3K–AKT–mTOR pathway was aberrantly activated in primary central nervous system lymphoma (PCNSL) and was associated with a poor prognosis. In addition, phosphorylated AKT was found to be widely expressed in 65.1% of patients with PCNSL [Citation72]. At the same time, George et al.(2005) detected AKT phosphorylation in 57% of patients with Anaplastic large cell lymphoma (ALCL) [Citation73]. Qingqing et al. (2012) reported that overexpression of phosphorylated AKT in peripheral T-cell lymphoma (PTCL) indicated shorter patient survival [Citation74]. Bruton’s tyrosine kinase (BTK) is important in the BCR signalling pathway [Citation75]. The application of BTK inhibitors has revolutionised the treatment of B-cell lymphoma [Citation76]; however, multiple mechanisms lead to resistance to BTK inhibitors. Kapoor et al. (2019) demonstrated activated AKT and downregulated PTEN and FOXO3a which causes Ibrutinib resistance in CLL, DLBCL, and MCL, which will explain how the opposite functioning will lead to the same outcome in this case [Citation77]. High expression of AKT directly phosphorylates downstream FOXO3a, leading to its uncontrolled presence in the cytoplasm. Studies have demonstrated that the poor prognosis of patients with MCL is closely related to the expression of phosphorylated FOXO3a [Citation78]. However, the function of PTEN is the opposite of that of PI3K, which inhibits AKT activation and blocks signalling [Citation79]. Therefore, understanding the relationship between the FOXO3a/PTEN/AKT signalling pathway and ibrutinib resistance is necessary. (3) Mutation or loss of regulatory factors of the PI3K–AKT signalling pathway can lead to activation of the pathway. Vallois et al. (2016) demonstrated that PDPK1, a positive regulator of the PI3K–AKT signalling pathway, was mutated or, in some cases, absent in 6% of patients with angioimmunoblastic T-cell lymphoma (AITL) [Citation80]. Germinal centre B-cell (GCB) DLBCL is haracterized by PTEN deficiency, which leads to enhanced activation of the PI3K–AKT–mTOR signalling pathway [Citation81]. GNA13 encodes the alpha subunit of G13 protein, which inhibits AKT phosphorylation and tumour proliferation in (GCB) DLBCL [Citation82]. Its mutation rate is 25% in CHL [Citation83]. B cell-specific Moloney murine leukaemia virus integration site 1 (Bmi-1) is an influential predictor of cancer invasion that promotes cell growth, enhances metastasis, and reduces apoptosis [Citation84]. BMI-1 is over-expressed in several tumours and plays an essential regulatory role in the malignant behaviour of tumours by activating the PI3K–AKT pathway [Citation85, Citation86]. Song et al. (2023) demonstrated that BMI-1 overexpression activated the PI3K–AKT signalling pathway and caused excessive proliferation of tumour cells in nude mouse models of DLBCL [Citation87]. High expression of c-myc contributes to the activity of downstream genes transcription and signalling pathways [Citation88]. The PI3k–Akt–mTOR pathway is one of the well-known signalling pathways downstream of c-myc. BL is an aggressive EBV-driven B-cell lymphoma that is characterized by translocation and rearrangement of c-myc [Citation89]. Sander et al. (2012) demonstrated that c-myc and the PI3K–AKT signalling pathway are activated in BL and are associated with a poorer prognosis [Citation90]. (4) mTOR activation is prevalent in lymphoma mainly owing to its upstream factors. mTOR is indirectly activated by AKT, a complex cell growth checkpoint that is affected by growth factor signalling, adenosine monophosphate content, and nutrient and oxygen accessibility [Citation67]. However, mTOR can act independently of AKT in some lymphomas, and experimental data indicate that suppression of mTOR is an effective strategy for the treatment of a broad spectrum of B-cell lymphomas [Citation91,Citation92] ().
Table 2. Distinct activation of the PI3K-AKT-mTOR pathway at different lymphoma diseases.
Early pan class I PI3K inhibitors were not clinically viable drugs due to significant off-target effects and solubilization problems [Citation93]. Recently, by modifying several inhibitors, the prospect of their clinical application has been restored. A phase I trial of SF 1126 in patients with advanced solid tumours and B-cell malignancies found stabilization in patients with chronic lymphocytic leukaemia (CLL) (50%; 2/4) and a 40% reduction in lymph node size after 1 cycle in patients with DLBCL [Citation94]. Perifosine is a first-generation Akt inhibitor [Citation95], and the combination of perifosine with the multikinase inhibitor sorafenib resulted in an ORR of 28% in a phase II trial for the treatment of recurrent Hodgkin lymphoma [Citation96]. Due to mild efficacy and moderate toxicity, there are no ongoing clinical trials evaluating the value of perifosine in patients with lymphoma. Rapamycin-like inhibitors, often labelled as ‘rapalogs’, are moderately active in lymphomas by variably inhibiting mTORC1.Tesirolimus has shown significant activity against relapsed mantle cell lymphoma when treated alone (ORR of 38% [Citation97]) and with rituximab (ORR of 59% [Citation98]). Based on these data, tesirolimus has received orphan drug approval in Europe for relapsed mantle cell lymphoma. Teicilomox has also demonstrated efficacy in other NHL subtypes, including follicular, SLL, and aggressive lymphomas [Citation99].
Targeting the PI3K/AKT/mTOR signalling pathway for the treatment of lymphoma is currently a hot topic in drug development. A new generation of PI3K inhibitors and AKT inhibitors are in early clinical stages, while a new generation of mTOR inhibitors has entered clinical trials, capable of blocking mTORC1 and mTORC2 and generating greater efficacy to avoid compensatory phosphorylation of Akt.
3. NF-KB signalling pathway
Aberrant activation of the NF-κB signalling pathway may be mediated by TLR/MyD88, BCR, CBM complex, and non-classical activation. NF-κB is a widely distributed and active eukaryotic transcription factor, which is a member of the REL protein family. The NF-κB family members found in mammalian cells include p50, p52, RelA (p65), RelB, and c-Rel, which form homodimers or heterodimers [Citation100]. NF-κB proteins share a Rel homology structural domain that contains a region where the REL interprotein dimerization site binds to the NF-κB inhibitor protein (IκB). In addition, this domain is involved in DNA recognition and nuclear localization [Citation101]. In the resting state, NF-κB is usually present in an inactive state and is bound to one of three inhibitory factors (IκBα, IκBβ, and IκBε) in the cytoplasm [Citation102]. In the presence of inflammatory factors; growth factors; and chemokines such as IL-1, IL-6, and TNF-α, which can activate NF-κB, IκB kinase (IKK) are phosphorylated by NF-κB-inducible kinase and degraded by the ubiquitin–proteasome pathway. When NF-κB and IKK complexes are depolymerized, their nuclear localization sequences are exposed and hence transported to the nucleus, where NF-κB binds to the DNA sequences of its target genes, initiates gene transcription, and stimulates the release of cytokines ().
Figure 3. The classical and alternative pathways of NF-KB signalling are mediated by a variety of receptors and signalling molecules, including TLR, CD30L, CD40L, RANKL, etc.; persistent EBV infection can also activate this pathway. In the classical NF-KB pathway, IKB binds to the P50-P65 heterodimer in the cytoplasm, preventing its entry into the nucleus. When receptor activation initiates adaptor proteins and signalling kinase responses, this will lead to activation of the IKB kinase complex(IKK), which phosphorylates IKB and leads to polyubiquitination and proteasomal degradation. This allows P50-P65 to be released into the nucleus to induce gene expression. In the alternative pathway, IKKα is activated by the upstream kinase NF-KB inducible kinase (NIK), which promotes the processing of P100-RelB into P52-RelB. Abnormal activation of the NF-KB pathway in lymphoma: 1. In MALT, CARD11, MALT1 and BCL10 are involved in the formation of the complex CBM, which is essential for the activation of NF-KB. 2. Meanwhile, overexpression of IAP2-MALT1 fusion proteins in MALT is also effective in activating NF-KB.
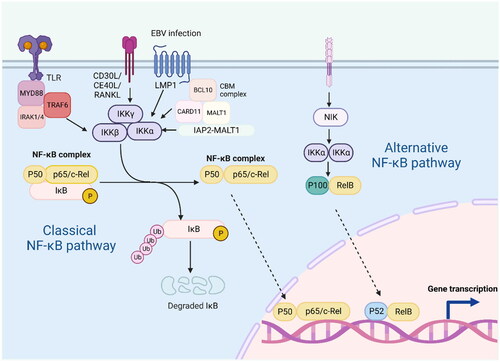
The NF-KB family plays an essential role in lymphocyte proliferation and survival. Different genetic disorders influence critical components of the NF-KB signalling pathway. NF-KB is aberrantly activated in various lymphoid malignancies, including DLBCL, HL, and CLL, MALT, PMBL and MCL [Citation32, Citation103–108]. Certain factors, such as continued infection (e.g. EBV infection), the pro-inflammatory immune microenvironment of cancer, and genetic mutations that alter key signalling pathway effectors, result in the activation of NF-KB activity in lymphoid malignancies. NF-kB is a key therapeutic target for many lymphomas. DLBCL development relies on the NF-KB signalling pathway, and the high frequency of mutations leading to NF-KB activation (such as CD79, CARD11 [Citation109], MYD88, or TNFAIP3) in activated B-cell (ABC) DLBCL suggests that the loss or activation of these genes promotes DLBCL development through aberrant activation of NF-KB [Citation110]. Recurrent oncogenic mutations of the MyD88 adaptor protein have been detected in various B-cell malignancies [Citation101]. Approximately 40% of ABC DLCBL biopsy specimens contain the MyD88 mutation, which represents the most commonly mutated oncogene in this tumour entity [Citation111]. Lu et al. (1991) reported that rearrangement or amplification of the Rel proto-oncogene is involved in the pathogenesis of DLBCL [Citation112]. In addition, the high incidence of genomic alterations in the NF-KB pathway is a key driver of HL pathogenesis. In HL, both typical and atypical NF-KB signalling pathways are activated, which may be promoted by the high expression of various TNF/death receptor family members, such as CD30, CD40, or RANK. Herishanu et al. (2011) revealed that LN-derived tumour cells are highly enriched in NF-KB target genes: CCND2, which is involved in cell cycle regulation; BCL2A1, which inhibits apoptosis; JUNB; DUSP2, which is involved in signal transduction; and CCL3; CCL4; and RGS1, which are involved in chemotaxis [Citation107]. In addition, activation of NF-KB in lymphoma may be attributed to the idiopathic translocation of gastric mucosa-associated lymphoid tissue (MALT), which involves MALT1 and BCL10. Both MALT1 and BCL10 are involved in the formation of the CARD11–BCL10–MALT1 (CBM) complex that activates the NF-KB signalling pathway [Citation113, Citation114]. Chromosomal translocations are specific to MALT, whose oncogenic product activates NF-KB. These include t(11;18) translocation that produces the c-IAP2–MALT1 fusion protein and t(14;18) and t(1;14) translocations that link the IgH locus enhancer to MALT1 and BCL10, respectively. In addition, excessive expression of the c-IAP2–MALT1 fusion protein can effectively activate NF-KB [Citation104]. Vilimas et al. found that most patients with T-cell acute lymphoblastic leukaemia (T-ALL) harboured mutations in the NOTCH1 gene. Constitutively active Notch1 activates NF-KB through transcription and the IKK complex, leading to tumour growth [Citation115]. Through a large-scale pharmacological analysis of more than 100 MCL cell lines, Rahal et al. found that the MCL model sensitive to BCR signalling inhibitors showed chronic activation of the BCR-driven NF-KB pathway, whereas the insensitive MCL model showed activation of the alternative NF-KB pathway. This finding indicates that the occurrence of MCL is associated with abnormal NF-KB signalling [Citation108].
Given that various genetic traits in lymphoma can activate NF-KB by deregulating different upstream signalling nodes, understanding how specific lesions drive NF-KB and the mechanisms that regulate lymphoma development may suggest novel strategies for targeting signalling pathways and improving the treatment of lymphoma. As major regulators of classical NF-κB signalling, IKK complexes, particularly IKKβ, constitute promising therapeutic targets. However, the critical and pleiotropic role of NF-κB in many physiological processes limits the therapeutic usefulness of classical IKK complexes. Therefore, targeting deregulated upstream pathways, such as chronically active BCR signalling (which drives constitutive NF-κB activation), may provide higher specificity for malignant cells and represent an attractive alternative for the treatment of lymphoid malignancies. The therapeutic efficacy of the BTK inhibitor ibrutinib in ABC DLBCL and other lymphoid cancers dependent on chronic BCR signalling demonstrates the validity of this concept [Citation116, Citation117]. MALT 1 proteinase inhibition may also be beneficial in patients with lymphoid malignancies dependent on constitutive CBM signalling or aberrant MALT 1 proteinase activity [Citation118, Citation119]. Meanwhile small molecule inhibitors targeting MYD88 have emerged as a potent therapeutic strategy for the treatment of MyD88-mutant lymphoid malignancies [Citation111, Citation120].
Conclusions
Investigation of the role of inflammatory pathways in the development of lymphoma has contributed to a deeper understanding of the pathogenesis of lymphoma. Inflammatory pathways directly or indirectly regulate the tumour immune microenvironment and play a crucial role in tumorigenesis. Although several studies have elucidated the functional roles of upstream and downstream target genes of many inflammatory pathways, their exact functions and regulatory mechanisms remain elusive. This review summarized the common oncogenic inflammatory pathways in lymphoma and the common molecular genetic events among them and discussed their role in the development of lymphoma and their regulatory effects on the tumour immune microenvironment. Lymphoma is a highly heterogeneous disease, and further investigation of its genetic and biological characteristics may help to develop more precise and individualized treatment options.
Authors’ contributions
A-R. Shi and Y-F. Jia participated in the conception and design of this study; F. Yun and L. Shi collected and analyzed the data; X. Liu contributed to interpretation of data; A-R. Shi contributed to drafting of the paper; X. Liu and Y-F. Jia revised it critically for intellectual content. All authors agreed to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Additional information
Funding
References
- Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of hemato-poietic and lymphoid tissues. 4th edition. Lyon (France): IARC Press. 2008.
- Thandra KC, Barsouk A, Saginala K, et al. Epidemiology of non-Hodgkin’s lymphoma. Med Sci. 2021;9(1):1. doi:10.3390/medsci9010005.
- Chu Y, Liu Y, Fang X, et al. The epidemiological patterns of non-Hodgkin lymphoma: global estimates of disease burden, risk factors, and temporal trends. Front Oncol. 2023;13:1059914. doi:10.3389/fonc.2023.1059914.
- Health Commission of the People’s Republic of China N. National guidelines for diagnosis and treatment of malignant lymphoma 2022 in China (English version). Chin J Cancer Res. 2022;34(5):425–13.
- Liu W, Liu J, Song Y, et al. Mortality of lymphoma and myeloma in China, 2004–2017: an observational study. J Hematol Oncol. 2019;12(1):22. doi:10.1186/s13045-019-0706-9.
- Matasar MJ, Zelenetz AD. Overview of lymphoma diagnosis and management. Radiol Clin North Am. 2008;46(2):175–198, vii. doi:10.1016/j.rcl.2008.03.005.
- Jiang M, Bennani NN, Feldman AL. Lymphoma classification update: T-cell lymphomas, Hodgkin lymphomas, and histiocytic/dendritic cell neoplasms. Expert Rev Hematol. 2017;10(3):239–249. doi:10.1080/17474086.2017.1281122.
- Aldinucci D, Gloghini A, Pinto A, et al. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumor growth and immune escape. J Pathol. 2010;221(3):248–263. doi:10.1002/path.2711.
- Bilotta MT, Antignani A, Fitzgerald DJ. Managing the TME to improve the efficacy of cancer therapy. Front Immunol. 2022;13(13):954992. doi:10.3389/fimmu.2022.954992.
- Liu WR, Shipp MA. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2017;2017(1):310–316. doi:10.1182/asheducation-2017.1.310.
- Jaeger A, Gambheer SMM, Sun X, et al. Activated granulocytes and inflammatory cytokine signaling drive T-cell lymphoma progression and disease symptoms. Blood. 2023;141(23):2824–2840. doi:10.1182/blood.2022015653.
- Patil K, Kuttikrishnan S, Khan AQ, et al. Molecular pathogenesis of cutaneous T cell lymphoma: role of chemokines, cytokines, and dysregulated signaling pathways. Semin Cancer Biol. 2022;86(Pt 3):382–399. doi:10.1016/j.semcancer.2021.12.003.
- Aittomäki S, Pesu M. Therapeutic targeting of the jak/STAT pathway. Basic Clin Pharmacol Toxicol. 2014;114(1):18–23. doi:10.1111/bcpt.12164.
- Hu X, Li J, Fu M, et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402.
- Fu XY. A transcription factor with SH2 and SH3 domains is directly activated by an interferon alpha-induced cytoplasmic protein tyrosine kinase(s). Cell. 1992;70(2):323–335. doi:10.1016/0092-8674(92)90106-m.
- Zhong Z, Wen Z, Darnell JE.Jr. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A. 1994;91(11):4806–4810. doi:10.1073/pnas.91.11.4806.
- Hou J, Schindler U, Henzel WJ, et al. An interleukin-4-induced transcription factor: IL-4 stat. Science. 1994;265(5179):1701–1706. doi:10.1126/science.8085155.
- Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci. 2000;25(10):496–502. doi:10.1016/s0968-0004(00)01624-8.
- Begitt A, Meyer T, van Rossum M, et al. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc Natl Acad Sci U S A. 2000;97(19):10418–10423. doi:10.1073/pnas.190318397.
- Kisseleva T, Bhattacharya S, Braunstein J, et al. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285(1–2):1–24. doi:10.1016/s0378-1119(02)00398-0.
- Chen X, Vinkemeier U, Zhao Y, et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93(5):827–839. doi:10.1016/s0092-8674(00)81443-9.
- Shuai K, Horvath CM, Huang LH, et al. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76(5):821–828. doi:10.1016/0092-8674(94)90357-3.
- Bousoik E, Montazeri Aliabadi H. Do we know Jack’ about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287. doi:10.3389/fonc.2018.00287.
- Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers . 2019;11(12):2002. doi:10.3390/cancers11122002.
- Zhao H, Wu L, Yan G, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. 2021;6(1):263.
- Xin P, Xu X, Deng C, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80:106210. doi:10.1016/j.intimp.2020.106210.
- O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. doi:10.1016/j.immuni.2012.03.014.
- Ritz O, Guiter C, Castellano F, et al. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood. 2009;114(6):1236–1242. doi:10.1182/blood-2009-03-209759.
- Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–3277. doi:10.1182/blood-2010-05-282780.
- Gaulard P, Harris NL, Pileri SA, et al. Primary mediastinal (thymic) large B-cell lymphoma. WHO classification of tumors of haematopoietic and lymphoid tissues. Geneva (Switzerland): WHO Press.2008:175–176.
- Stein H, von Wasielewski R, Poppema S, et al. Nodular sclerosis classical Hodgkin lymphoma. WHO classification of tumors of haematopoietic and lymphoid tissues. Geneva (Switzerland): WHO Press; 2008:249–250.
- Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical hodgkin lymphoma. Blood. 2003;102(12):3871–3879. doi:10.1182/blood-2003-06-1841.
- Phillips D, Matusiak M, Gutierrez BR, et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nat Commun. 2021;12(1):6726. doi:10.1038/s41467-021-26974-6.
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92(10):1085–1102. doi:10.1002/ajh.24876.
- Mehta-Shah N, Horwitz SM, Ansell S, et al. NCCN guidelines insights: primary cutaneous lymphomas, version 2.2020. J Natl Compr Canc Netw. 2020;18(5):522–536. doi:10.6004/jnccn.2020.0022.
- Tensen CP, Quint KD, Vermeer MH. Genetic and epigenetic insights into cutaneous T-cell lymphoma. Blood. 2022;139(1):15–33. doi:10.1182/blood.2019004256.
- Gluud M, Pallesen EMH, Buus TB, et al. Malignant T cells induce skin barrier defects through cytokine-mediated JAK/STAT signaling in cutaneous T-cell lymphoma. Blood. 2023;141(2):180–193. doi:10.1182/blood.2022016690.
- Nestle FO, Di Meglio P, Qin JZ, et al. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–691. doi:10.1038/nri2622.
- Pendaries V, Malaisse J, Pellerin L, et al. Knockdown of filaggrin in a three-dimensional reconstructed human epidermis impairs keratinocyte differentiation. J Invest Dermatol. 2014;134(12):2938–2946. doi:10.1038/jid.2014.259.
- Kezic S, Kemperman PM, Koster ES, et al. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J Invest Dermatol. 2008;128(8):2117–2119. doi:10.1038/jid.2008.29.
- Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–597. doi:10.1158/2159-8290.CD-12-0028.
- Manshouri T, Quintás-Cardama A, Nussenzveig RH, et al. The JAK kinase inhibitor CP-690,550 suppresses the growth of human polycythemia vera cells carrying the JAK2V617F mutation. Cancer Sci. 2008;99(6):1265–1273. doi:10.1111/j.1349-7006.2008.00817.x.
- Rivera-Munoz P, Laurent AP, Siret A, et al. Partial trisomy 21 contributes to T-cell malignancies induced by JAK3-activating mutations in murine models. Blood Adv. 2018;2(13):1616–1627. doi:10.1182/bloodadvances.2018016089.
- Cornejo MG, Kharas MG, Werneck MB, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood. 2009;113(12):2746–2754. doi:10.1182/blood-2008-06-164368.
- Gozgit JM, Bebernitz G, Patil P, et al. Effects of the JAK2 inhibitor, AZ960, on pim/BAD/BCL-xL survival signaling in the human JAK2 V617F cell line SET-2. J Biol Chem. 2008;283(47):32334–32343. doi:10.1074/jbc.M803813200.
- Tiacci E, Ladewig E, Schiavoni G, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood. 2018;131(22):2454–2465. doi:10.1182/blood-2017-11-814913.
- Mathas S, Hartmann S, Küppers R. Hodgkin lymphoma: pathology and biology. Semin Hematol. 2016;53(3):139–147. doi:10.1053/j.seminhematol.2016.05.007.
- Morin RD, Assouline S, Alcaide M, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22(9):2290–2300. doi:10.1158/1078-0432.CCR-15-2123.
- Yildiz M, Li H, Bernard D, et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125(4):668–679. doi:10.1182/blood-2014-06-582650.
- Dubois S, Viailly PJ, Mareschal S, et al. Next-Generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22(12):2919–2928. doi:10.1158/1078-0432.CCR-15-2305.
- Scheeren FA, Diehl SA, Smit LA, et al. IL-21 is expressed in hodgkin lymphoma and activates STAT5: evidence that activated STAT5 is required for hodgkin lymphomagenesis. Blood. 2008;111(9):4706–4715. doi:10.1182/blood-2007-08-105643.
- Zhang L, Yang J, Qian J, et al. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood. 2012;120(18):3783–3792. doi:10.1182/blood-2012-04-424630.
- Kuruvilla J, MacDonald DA, Kouroukis CT, et al. Salvage chemotherapy and autologous stem cell transplantation for transformed indolent lymphoma: a subset analysis of NCIC CTG LY12. Blood. 2015;126(6):733–738. doi:10.1182/blood-2015-01-622084.
- Ramis-Zaldivar JE, Gonzalez-Farre B, Nicolae A, et al. MAPK and JAK-STAT pathways dysregulation in plasmablastic lymphoma. Haematologica. 2021;106(10):2682–2693. doi:10.3324/haematol.2020.271957.
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2018;68(2):116–132. doi:10.3322/caac.21438.
- Diaz T, Navarro A, Ferrer G, et al. Lestaurtinib inhibition of the Jak/STAT signaling pathway in hodgkin lymphoma inhibits proliferation and induces apoptosis. PLOS One. 2011;6(4):e18856. doi:10.1371/journal.pone.0018856.
- Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019;20(9):515–534. doi:10.1038/s41580-019-0129-z.
- Jafari M, Ghadami E, Dadkhah T, et al. PI3k/AKT signaling pathway: erythropoiesis and beyond. J Cell Physiol. 2019;234(3):2373–2385. doi:10.1002/jcp.27262.
- Xu Z, Han X, Ou D, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–587. doi:10.1007/s00253-019-10257-8.
- Zhao Y, Qian Y, Sun Z, et al. Role of PI3K in the progression and regression of atherosclerosis. Front Pharmacol. 2021;12:632378. doi:10.3389/fphar.2021.632378.
- Glaviano A, Foo ASC, Lam HY, et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138. doi:10.1186/s12943-023-01827-6.
- Nepstad I, Hatfield KJ, Grønningsæter IS, et al. The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci. 2020;21(8):2907. doi:10.3390/ijms21082907.
- Tewari D, Patni P, Bishayee A, et al. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: a novel therapeutic strategy. Semin Cancer Biol. 2022;80:1–17. doi:10.1016/j.semcancer.2019.12.008.
- Cornejo MG, Mabialah V, Sykes SM, et al. Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood. 2011;118(5):1264–1273. doi:10.1182/blood-2011-01-328567.
- Abubaker J, Bavi PP, Al-Harbi S, et al. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia. 2007;21(11):2368–2370. doi:10.1038/sj.leu.2404873.
- Psyrri A, Papageorgiou S, Liakata E, et al. Phosphatidylinositol 3’-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res. 2009;15(18):5724–5732. doi:10.1158/1078-0432.CCR-08-3215.
- Westin JR. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin Lymphoma Myeloma Leuk. 2014;14(5):335–342. doi:10.1016/j.clml.2014.01.007.
- Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8(10):1153–1160. doi:10.1038/nm761.
- Héron-Milhavet L, Franckhauser C, Rana V, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol. 2006;26(22):8267–8280. doi:10.1128/MCB.00201-06.
- Georgakis GV, Li Y, Rassidakis GZ, et al. Inhibition of the phosphatidylinositol-3 kinase/Akt promotes G1 cell cycle arrest and apoptosis in Hodgkin lymphoma. Br J Haematol. 2006;132(4):503–511. doi:10.1111/j.1365-2141.2005.05881.x.
- Uddin S, Hussain AR, Siraj AK, et al. Role of phosphatidylinositol 3’-kinase/AKT pathway in diffuse large B-cell lymphoma survival. Blood. 2006;108(13):4178–4186. doi:10.1182/blood-2006-04-016907.
- Zhang X, Wu Y, Sun X, et al. The PI3K/AKT/mTOR signaling pathway is aberrantly activated in primary Central nervous system lymphoma and correlated with a poor prognosis. BMC Cancer. 2022;22(1):190. doi:10.1186/s12885-022-09275-z.
- Rassidakis GZ, Feretzaki M, Atwell C, et al. Inhibition of Akt increases p27Kip1 levels and induces cell cycle arrest in anaplastic large cell lymphoma. Blood. 2005;105(2):827–829. doi:10.1182/blood-2004-06-2125.
- Cai Q, Deng H, Xie D, et al. Phosphorylated AKT protein is overexpressed in human peripheral T-cell lymphomas and predicts decreased patient survival. Clin Lymphoma Myeloma Leuk. 2012;12(2):106–112. doi:10.1016/j.clml.2011.12.002.
- Woyach JA, Bojnik E, Ruppert AS, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood. 2014;123(8):1207–1213. doi:10.1182/blood-2013-07-515361.
- Wang H, Zhang W, Yang J, et al. The resistance mechanisms and treatment strategies of BTK inhibitors in B-cell lymphoma. Hematol Oncol. 2021;39(5):605–615. doi:10.1002/hon.2933.
- Kapoor I, Li Y, Sharma A, et al. Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies. Cell Death Dis. 2019;10(12):924. doi:10.1038/s41419-019-2158-0.
- Obrador-Hevia A, Serra-Sitjar M, Rodríguez J, et al. The tumour suppressor FOXO3 is a key regulator of mantle cell lymphoma proliferation and survival. Br J Haematol. 2012;156(3):334–345. doi:10.1111/j.1365-2141.2011.08951.x.
- Bu L, Wang H, Pan JA, et al. PTEN suppresses tumorigenesis by directly dephosphorylating Akt. Signal Transduct Target Ther. 2021;6(1):262.
- Vallois D, Dobay MP, Morin RD, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128(11):1490–1502. doi:10.1182/blood-2016-02-698977.
- Pfeifer M, Grau M, Lenze D, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(30):12420–12425. doi:10.1073/pnas.1305656110.
- Xia Z, Zhang X, Liu P, et al. GNA13 regulates BCL2 expression and the sensitivity of GCB-DLBCL cells to BCL2 inhibitors in a palmitoylation-dependent manner. Cell Death Dis. 2021;12(1):54. doi:10.1038/s41419-020-03311-1.
- Desch AK, Hartung K, Botzen A, et al. Genotyping circulating tumor DNA of pediatric hodgkin lymphoma. Leukemia. 2020;34(1):151–166. doi:10.1038/s41375-019-0541-6.
- Li Y, Tian Z, Tan Y, et al. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol Cancer. 2020;19(1):109. doi:10.1186/s12943-020-01229-y.
- Zaczek A, Jóźwiak P, Ciesielski P, et al. Relationship between polycomb-group protein BMI-1 and phosphatases regulating AKT phosphorylation level in endometrial cancer. J Cell Mol Med. 2020;24(2):1300–1310. doi:10.1111/jcmm.14782.
- Yang XX, Ma M, Sang MX, et al. BMI-1 suppression increases the radiosensitivity of oesophageal carcinoma via the PI3K/Akt signaling pathway. Oncol Rep. 2018;39(2):667–678.
- Song Y, Yang Z, Li W, et al. The study of the mechanism of BMI-1 promoting the lymphoma growth in nude mice via activating PI3K/AKT signaling pathway. Modern Oncol. 2023;31(06):985–991.
- Wang C, Zhang J, Yin J, et al. Alternative approaches to target myc for cancer treatment. Signal Transduct Target Ther. 2021;6(1):117.
- López C, Burkhardt B, Chan JKC, et al. Burkitt lymphoma. Nat Rev Dis Primers. 2022;8(1):78. doi:10.1038/s41572-022-00404-3.
- Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in burkitt lymphomagenesis. Cancer Cell. 2012;22(2):167–179. doi:10.1016/j.ccr.2012.06.012.
- Dal Col J, Zancai P, Terrin L, et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111(10):5142–5151. doi:10.1182/blood-2007-07-103481.
- Wlodarski P, Kasprzycka M, Liu X, et al. Activation of mammalian target of rapamycin in transformed B lymphocytes is nutrient dependent but independent of Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase, insulin growth factor-I, and serum. Cancer Res. 2005;65(17):7800–7808. doi:10.1158/0008-5472.CAN-04-4180.
- Yap TA, Garrett MD, Walton MI, et al. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8(4):393–412. doi:10.1016/j.coph.2008.08.004.
- Mahadevan D, Chiorean EG, Harris WB, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. 2012;48(18):3319–3327. doi:10.1016/j.ejca.2012.06.027.
- Kondapaka SB, Singh SS, Dasmahapatra GP, et al. A novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2(11):1093–1103.
- Guidetti A, Viviani S, Marchiano A, et al. Dual targeted therapy with the AKT inhibitor perifosine and the multikinase inhibitor sorafenib in patients with relapsed/refractory lymphomas: final results of a phase II trial. ASH Annual Meeting Abstracts. 2012;1203679.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347–5356. doi:10.1200/JCO.2005.13.466.
- Kumar S, Mikhael JR, LaPlant BR, et al. Phase I trial of a novel combination of an HDAC inhibitor (LBH589) and an mTOR inhibitor (RAD001) in lymphoid and plasma cell malignancies. ASH Annual Meeting Abstracts. 2011;118:2682.
- Smith SM, van Besien K, Karrison T, et al. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: the university of Chicago phase II consortium. J Clin Oncol. 2010;28(31):4740–4746. doi:10.1200/JCO.2010.29.2813.
- Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflamm Res. 2018;11:407–419. doi:10.2147/JIR.S140188.
- Grondona P, Bucher P, Schulze-Osthoff K, et al. NF-κB activation in lymphoid malignancies: genetics, signaling, and targeted therapy. Biomedicines. 2018;6(2):38. doi:10.3390/biomedicines6020038.
- Mitchell JP, Carmody RJ. NF-κB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. 2018;335:41–84.
- Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109–a000109. doi:10.1101/cshperspect.a000109.
- Lim KH, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunol Rev. 2012;246(1):359–378. doi:10.1111/j.1600-065X.2012.01105.x.
- Bargou RC, Leng C, Krappmann D, et al. High-level nuclear NF-kappa B and oct-2 is a common feature of cultured hodgkin/Reed-Sternberg cells. Blood. 1996;87(10):4340–4347. doi:10.1182/blood.V87.10.4340.bloodjournal87104340.
- Davis RE, Brown KD, Siebenlist U, et al. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861–1874. doi:10.1084/jem.194.12.1861.
- Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574. doi:10.1182/blood-2010-05-284984.
- Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. doi:10.1038/nm.3435.
- Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676–1679. doi:10.1126/science.1153629.
- Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–721. doi:10.1038/nature07968.
- Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi:10.1038/nature09671.
- Lu D, Thompson JD, Gorski GK, et al. Alterations at the rel locus in human lymphoma. Oncogene. 1991;6(7):1235–1241.
- Bertin J, Wang L, Guo Y, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276(15):11877–11882. doi:10.1074/jbc.M010512200.
- Gaide O, Favier B, Legler DF, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3(9):836–843. doi:10.1038/ni830.
- Vilimas T, Mascarenhas J, Palomero T, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13(1):70–77. doi:10.1038/nm1524.
- Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92.doi:10.1038/nature08638.
- Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21(8):922–926. doi:10.1038/nm.3884.
- Dai B, Grau M, Juilland M, et al. B-cell receptor-driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood. 2017;129(3):333–346. doi:10.1182/blood-2016-05-718775.
- Hailfinger S, Lenz G, Ngo V, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2009;106(47):19946–19951. doi:10.1073/pnas.0907511106.
- Kelly PN, Romero DL, Yang Y, et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J Exp Med. 2015;212(13):2189–2201. doi:10.1084/jem.20151074.