Abstract
Small heat shock proteins (small HSPs) are molecular chaperones that protect cells against stress by assisting in the correct folding of denatured proteins and thus prevent aggregation of misfolded proteins. Small HSPs also modulate apoptotic pathways by interacting with components of programmed cell death. Furthermore, some small HSPs interact with the cytoskeleton to assist in spatial organization and dynamics of its structural elements. The role of small HSPs has been studied in many disorders, including neurodegenerative disease. Recently, mutations in HSPB1 (HSP27) and HSPB8 (HSP22), two members of the small HSP superfamily, have been associated with inherited peripheral neuropathies. In this review, we will summarize the current knowledge of small HSPs, in particular HSPB1 and HSPB8, and discuss their role in health and disease.
Introduction
Heat shock proteins (HSP) act as molecular chaperones and protect intracellular components during stress conditions. Even in unstressed cells, HSPs are constitutively expressed to properly fold and assemble polypeptides. The name ‘heat shock protein’ in fact refers to the rapid induction of these proteins in response to elevated temperatures, although other stress stimuli (e.g. oxidative stress, heavy metals and ischemia injury) are also capable of inducing expression of HSPs. HSPs are classified according to their molecular weight and currently five major classes are recognized: HSP100, HSP90, HSP70, HSP60 and the small heat shock proteins (small HSPs), which have a monomer size between 12 and 43 kDa. In this review we will focus on the small HSPs. In vitro these small HSPs have a chaperone‐like activity, i.e. the capacity to interact with partially denatured proteins and to prevent protein misfolding and aggregation. They are also involved in diverse cellular activities such as modulation of actin and intermediate filament dynamics Citation1, Citation2, cellular growth and differentiation Citation3, Citation4 and apoptosis Citation5.
Genes encoding small HSPs are found in all organisms, from bacteria to vertebrates. In addition, all members of the small HSP superfamily share a conserved C‐terminal α‐crystallin domain, containing 80–100 amino acid residues, a more variable N‐terminal sequence and a short variable C‐terminal tail. The combination of a conserved domain and their universal presence, indicate a crucial, evolutionary role in cellular homeostasis. In human, the ten genes encoding small HSPs are named HSPB1 to HSPB10 according to the HUGO Gene Nomenclature Committee Citation6, Citation7. Recently, five novel small HSPs were identified in other vertebrates, HSPB11–15, but these do not have a mammalian orthologue Citation8. In retrospect, the first members of the small HSP superfamily, αA‐crystallin/HSPB4 and αB‐crystallin/HSPB5, were identified more than a century ago as ‘α‐crystallins’ Citation9. Only in 1982 the relationship of these proteins with the small HSP superfamily was recognized Citation10. HSPB4 is almost exclusively expressed in the lens and is not heat‐inducible, while HSPB5 is distributed ubiquitously and is heat‐inducible Citation11. HSPB1 (HSP27), a heat‐inducible small HSP that was identified 20 years ago, is ubiquitously expressed in human tissues and interacts with HSPB8, another small HSP Citation12. HSPB8, also known as HSP22, is a heat‐inducible small HSP, expressed in various tissues including muscle, motor and sensory neurons Citation13–16. The remaining small HSPs will not be discussed in detail since so far they have not been implicated in human disorders.
During the last decade, the involvement of small HSPs has been studied in many disorders, including neurodegenerative diseases. The first studies focused on the potential protective effect of upregulation of HSP expression. Increased levels of wild‐type small HSPs, especially HSPB5 and HSPB1, were indeed found in individuals with neurodegenerative diseases, such as Alzheimer Citation17, Parkinson Citation18, Alexander disease Citation19 and amyotrophic lateral sclerosis (ALS) Citation20. These neurodegenerative disorders are all marked by the deposition of improperly folded proteins, inclusion bodies or plaques in the nervous system.
On the other hand, mutations in four small HSPs have been associated with autosomal dominant and recessive hereditary disorders. Mutations in the crystallin genes, HSPB4 and HSPB5, are associated with congenital cataract and desmin‐related myopathy Citation21, Citation22. Mutations in the interacting small HSPs, HSPB1 and HSPB8, have been shown to cause distal hereditary motor neuropathy (distal HMN type II) and Charcot‐Marie‐Tooth disease type 2 (CMT2) () Citation23–25. It is not clear what the underlying pathological mechanism is in these disorders: misfolding and aggregation of the mutant HSP, a defective cytoprotective activity leading to misfolding and aggregation of other proteins, a dysfunction in cell death pathways, or a combination of these effects.
Table I. Mutations in HSPB1 and HSPB8 and the associated phenotypes.
Because mutations in HSPB1 and HSPB8 were only recently identified as a cause of inherited peripheral neuropathies, we will concentrate on these two small HSPs and discuss how mutations in these genes affect motor and sensory neurons.
Key messages
Small heat shock proteins (small HSPs) have cytoprotective roles including chaperone‐like activity, anti‐apoptotic activity and preservation of cytoskeletal elements.
The neuroprotective role of small HSPs has been studied in many disorders, including neurodegenerative disease.
Recently, mutations in HSPB1 (HSP27) and HSPB8 (HSP22), two members of the small HSP superfamily, have been associated with inherited peripheral neuropathies.
HSPB1 and HSPB8 mutations in inherited peripheral neuropathies
Distal HMN, also known as hereditary distal spinal muscular atrophy (distal SMA) is a pure motor disorder of the peripheral nervous system (PNS) Citation26. Distal HMN is clinically characterized by progressive weakness and atrophy of the muscles in the lower limbs. Later on weakness is also present in the distal upper limbs. In a related inherited peripheral neuropathy, CMT2, sensory neurons are also involved. On clinical examination, distal HMN patients are hard to distinguish from CMT2 patients, because in CMT2 patients sensory symptoms are often absent. Therefore electrophysiological examinations are essential to confirm the diagnosis of either distal HMN, where only motor neurons are involved, or CMT2, where motor and sensory neurons are involved Citation27. Both disorders are clinically and genetically heterogeneous Citation28, Citation29. Recently, missense mutations in HSPB1 and HSPB8 have been associated with distal HMN and CMT2 () Citation23–25. Initially two distinct missense mutations (K141N and K141E), involving the same amino acid in the α‐crystallin domain of HSPB8 were found in four distal HMN type II families. Subsequently, the heterozygous amino acid change (K141N) in HSPB8 was also reported in a large Chinese CMT2 family (CMT type 2L) which was previously linked to the same chromosomal region as distal HMN type II Citation25, Citation30. These observations raise two important points. First, the same HSPB8 missense mutation (K141N) is associated with two distinct phenotypes, i.e. distal HMN and CMT2. Secondly, all reported pathogenic mutations so far are missense mutations that target the same amino acid. The conserved K141 residue in HSPB8 corresponds to R116 and R120 of the HSPB4 and HSPB5 proteins respectively (). Interestingly, these arginine residues are mutated in patients with congenital cataract and desmin‐related myopathy Citation21, Citation22. Biochemical analysis of the R116C and R120G missense mutations in HSPB4 and HSPB5 shows significant changes in the secondary and tertiary structure of these small HSPs, a defective chaperone‐like activity and a higher aggregate size Citation31. The R116C mutation in HSPB4 also reduces the protective ability in lens epithelial cell apoptosis Citation32. Members of the small HSP superfamily interact with each other and changes in these interactions may be a culprit mechanism in pathological conditions. The interaction between mutant HSPB4 (R116C) and wild‐type HSPB1 and HSPB5 is increased, whereas the interaction with HSPB4 wild‐type proteins shows little change Citation33. Co‐immunoprecipitation studies demonstrated also that mutant HSPB8 (K141N/E) has an increased interaction with HSPB1 and thus might interfere with the activity of HSPB1 Citation23. These studies underline the importance of the preservation of a positively charged amino acid at that position in these small HSPs. In vitro studies have also shown that mutant HSPB8 promotes the formation of cytoplasmatic and perinuclear aggregates, which correlated with a significant decrease in neuronal cell viability Citation23. Aggregation of desmin filaments is also induced by mutant HSPB5 (R120G) Citation34. Accumulation of these misfolded proteins can have deleterious effects on neuronal cell function by disrupting the axonal transport and can ultimately lead to cell death ().
Figure 1 Structure‐based sequence alignments of HSPB1 and HSPB8 orthologues and homologues HSPB4 and HSPB5. The N‐terminal WD/EPF and SRLDQF/AFG domain, the α‐crystallin domain and the C‐terminal IXI/V domain are indicated. The peripheral neuropathy‐associated mutations are boxed in grey. The cytochrome C binding motifs are indicated by a dotted line.
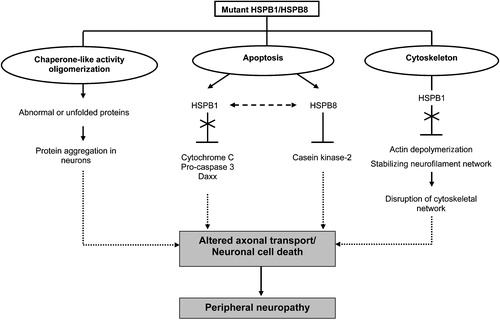
Figure 2 Suggested pleiotropic actions of mutant HSPB1 and HSPB8. Mutations in HSPB1 and HSPB8 can interfere with the chaperone‐like activity, causing insoluble cellular aggregates and subsequent induction of neuronal cell death. HSPB1 and HSPB8 regulate different apoptotic pathways. Mutant HSPB1 can no longer bind cytochrome C, pro‐caspase 3 and Daxx and is thus unable to prevent apoptotic cell death. Mutations in HSPB8 may result in enhanced apoptosis by inhibiting the pro‐survival activity of casein kinase‐2 or by negatively influencing the anti‐apoptotic activity of its interacting partner, HSPB1. Furthermore, mutant HSPB1 is able to disrupt cytoskeletal functions and neurofilament assembly, resulting in neuronal cell death or altered axonal transport. Ultimately, these pathological mechanisms will cause peripheral neuropathy in humans.
Not entirely unexpected, missense mutations in HSPB1, the molecular partner of HSPB8, were also associated with distal HMN and CMT type 2F Citation24. Four mutations in HSPB1 target the highly conserved α‐crystallin domain (R127W, S135F, R136W and T151I) and one is positioned in the C‐terminal part (P182L) (; ). Experimental studies with HSPB5 showed that mutations in the core α‐crystallin domain decrease the chaperone function Citation35. The HSPB1 mutations found in distal HMN and CMT2F patients may therefore interfere with the chaperone activity. The C‐terminus of several small HSPs contains a conserved IXI/V motif (). This motif is relevant for inter‐subunit interactions and oligomerization, either with regions in the α‐crystallin domain or with IXI/V motifs on other monomers Citation36. Possibly, the C‐terminal missense mutation (P182L) in HSPB1 interferes with subunit‐interactions of small HSPs, which are important to maintain the oligomeric state of the protein. Furthermore, this IXI/V motif is critical in the chaperone‐like activity of several bacterial small HSPs. Deletion of this motif or mutations in this motif, result in a complete loss of chaperone activity and the truncated protein was unable to form oligomers Citation37. In addition, a site‐directed mutation of P160 in HSPB5, which corresponds to P182 in HSPB1, also influences the chaperone function Citation35. These effects may contribute to the reduced cell viability of neuronal cells seen in transient transfection studies with mutated HSPB1 Citation24.
Cellular and cytoprotective properties of HSPB1 and HSPB8
Under normal conditions HSPB1, as most small HSPs, forms large oligomers consisting of about 24 subunits. These oligomeric complexes are formed by dimeric building blocks composed of small HSP monomers. The well conserved N‐terminal SRLFDQFFG motif determines the oligomeric size of small HSPs and contributes to their structural stability () Citation38. A unique cysteine residue in Hspb1 (C141) regulates the formation of dimers Citation39. Hspb1 with a mutation at this cysteine residue showed decreased ability to multimerize, and was less efficient in inhibiting staurosporine‐induced apoptosis Citation39. Interestingly, the cysteine residue, corresponding to C137 in HSPB1, is located next to the amino acids S135 and R136, which are mutated in individuals with a motor and sensory neuropathy Citation24. Mutations located near this functionally important cysteine residue, possibly interfere with oligomerization. The oligomeric structure of HSPB8 is not clear. An in vitro study showed that Hspb8 exists as a monomer in contrast to other small HSPs Citation40, however in other studies, HSPB8 predominantly formed dimers Citation15, Citation41 and high molecular mass complexes in the heart Citation42. The oligomerization of mammalian small HSPs is regulated by phosphorylation Citation1. Mitogen activating proteins (MAP), MAPKAP kinase 2 and 3, which are activated by p38 MAP kinase, are the main kinases involved in the phosphorylation of small HSPs. Stress‐induced phosphorylation and oligomerization may thus play an important role in the biological activities of small HSPs Citation43, Citation44.
We will now summarize the three main cytoprotective functions of small HSPs: 1) the chaperone‐like activity, 2) the capacity to interfere with cellular death pathways and 3) the role in stabilizing the cytoskeleton ().
HSPB1 and HSPB8 act as molecular chaperones
Small HSPs contribute to protection of the cell against stress by their chaperone‐like activity, i.e. their capacity to bind non‐native proteins. In vitro studies have demonstrated that mammalian small HSPs recognize misfolded proteins and prevent intracellular protein aggregation by keeping them in a soluble phase Citation45. The in vitro molecular chaperone activity of HSPB1 and HSPB8 has been extensively documented Citation34, Citation40, Citation41, Citation44, Citation46. They prevented aggresome formation in cells expressing mutant HSPB5 (R120G) by forming complexes with misfolded HSPB5 Citation34. Interestingly, in contrast to HSPB1, HSPB8 did not remain associated with the mutant HSPB5 complexes. This suggests that HSPB8 is involved in protein folding, and therefore can be considered as a true chaperoning molecule Citation34.
Several studies demonstrated an important, but not sufficient role of the α‐crystallin domain in the chaperone‐activity of small HSPs Citation35, since additional parts of the protein are required, such as the N‐terminal proline‐phenylalanine‐rich region containing a WD/EPF (single letter amino acid code) motif in HSPB1 and the C‐terminal region () Citation47, Citation48. The chaperone‐like activity of small HSPs, as documented in in vitro studies, may of course be highly relevant for their role in human disorders. However this activity still needs to be confirmed by in vivo studies Citation45.
HSPB1 and HSPB8 act as modulators of apoptosis
The protective effect of small HSPs is also related to their interference with apoptotic pathways. HSPB1 prevents the formation of apoptosomes and prevents cell death triggered by various stimuli. HSPB1 maintains the redox status and mitochondrial stability in the cell and prevents apoptosis due to reactive oxygen species Citation44, Citation49. It also inhibits apoptosis by direct binding to molecular components of cell death pathways. HSPB1 binds to both cytochrome C, when it is released from the mitochondria to the cytosol, and to pro‐caspase‐3, thus preventing the activation of the caspase cascade () Citation50, Citation51. The cysteine residue at position 137, as well as the N‐terminal region, seem to be essential for binding to cytochrome C () Citation50. HSPB1 also affects the caspase‐independent apoptotic pathway, by its interference in the translocation of Daxx to the membrane and its interaction with Fas Citation52. Daxx is a nuclear protein that translocates to the membrane during Fas‐mediated caspase‐independent apoptosis.
Another function of HSPB1 related to cell death, is its role in proteasome‐mediated degradation of selected proteins. HSPB1 is able to interact with specific components of the proteasome and to bind ubiquitin, thus facilitating protein degradation Citation53. The interaction between HSPB1 and ubiquitin explains the co‐localization with ubiquitinated proteins in cytoplasmatic inclusions that characterize some neurodegenerative diseases.
In contrast to the anti‐apoptotic activity of most small HSPs, a pro‐apoptotic activity was assigned to HSPB8 when forced to express in tumor cell lines Citation54. HSPB8 triggered apoptosis is caspase‐ and p38 MAP kinase‐dependent and cell‐type specific since not all cell‐types overexpressing HSPB8 showed an enhanced apoptosis Citation54. This is well documented in the heart were HSPB8 plays a dual role; at low doses it induces myocardial hypertrophy through the activation of the Akt or protein kinase B (Akt/PKB) pathway, while at high doses it has a pro‐apoptotic activity via protein kinase‐dependent mechanisms Citation55. HSPB8 also interacts with casein kinase‐2 (CK‐2), which has anti‐apoptotic effects. It is speculated that HSPB8 promotes apoptosis by inhibition of the CK‐2 activity in cardiac myocytes Citation55. Since HSPB8 interacts with HSPB1 and other small HSPs, mutant HSPB8 may induce apoptosis by disturbing the anti‐apoptotic effect of other small HSPs Citation23, Citation42. Although the apoptotic activity of HSPB8 in motor neurons is unknown, both HSPB8 and HSPB1 associate with the Akt/PKB kinase pathway, a major signaling pathway in non‐neuronal and neuronal cells for growth and survival Citation56, Citation57. This pathway is also activated in spinal motor neurons after nerve injury, wherein HSPB1 plays a role in promoting cell survival Citation56.
HSPB1 and HSPB8 act as protectors of the cytoskeleton
A cytoprotective function of HSPB1 is also to regulate cytoskeletal dynamics. The cytoskeleton maintains the shape of the cell by modulating the spatial arrangement and dynamics of its structural elements, consisting of microtubules, intermediate filaments and actin microfilaments. During stress, the integrity of this cytoskeletal network is disrupted by disorganization and aggregation of the filament system. Abnormalities in cytoskeletal organization and aggregates are frequent histopathological hallmarks in neurodegenerative disorders. HSPB1 and microfilaments interact to prevent disruption of the cytoskeleton during exposure to stress stimuli Citation2. Although the molecular mechanism by which HSPB1 stabilizes the actin network is not fully understood, an inhibition of actin polymerization was demonstrated Citation58. The depolymerization of actin is regulated by the structural organization of HSPB1 and its p38 MAP kinase‐mediated phosphorylation Citation59. Unphosphorylated HSPB1 monomers effectively block polymerization, whereas phosphorylated monomers and unphosphorylated oligomers are not able to prevent actin polymerization Citation58. The interaction between HSPB1 and actin, and the related depolymerization effect were assigned to amino acid sequences at the N‐terminus of HSPB1 Citation60.
Besides the interaction with actin microfilaments, HSPB1 also associates with intermediate filaments. In vitro studies show that this interaction controls filament‐filament interactions, which are important for cell survival by preventing intermediate filament aggregation Citation61. The fact that mutant HSPB1 (S135F) results in disruption of the cellular neurofilament (NF‐L) network, provides evidence for an essential role of HSPB1 in regulating this dynamic network Citation24. Furthermore, the R120G mutation in HSPB5, is also associated with impaired neurofilament assembly, resulting in pathological aggregates of desmin filaments Citation34. These findings underscore the functional relationship between small HSPs and the cytoskeleton, and link several neuronal diseases with intermediate filaments. Interestingly, mutations in the neurofilament light chain gene (NEFL) cause another variant of CMT2 (CMT2E) Citation62 and mutations in the peripherin gene (PRPH) are associated with ALS Citation63. At present, HSPB8 has not been shown to interact directly with cytoskeletal elements, however since small HSPs operate in association with each other, it is possible that HSPB8 does interact with microfilaments and intermediate filaments via its molecular partner HSPB1. The missense mutations in HSPB1 and HSPB8 in distal HMN and CMT2 could therefore interfere either directly or indirectly with cytoskeletal functions and affect axonal transport in motor and sensory neurons (). Of note is that mutations in dynamin 2 (DNM2) and dynactin (DCTN1), proteins that are involved in axonal transport, result in peripheral neuropathy Citation64, Citation65.
Neuroprotective role of HSPB1
HSPB1 is expressed in the PNS and might be important for axonal outgrowth, since it is induced in Schwann cells and regenerated axons Citation66, Citation67. Furthermore, expression of HSPB1 is upregulated after damage to adult peripheral sensory and motor neurons Citation66, Citation68, Citation69. In addition, exogenous delivery of HSPB1 is able to rescue injured sensory, sympathetic and motor neurons in vitro and in vivoCitation69, Citation70. This neuroprotective activity of HSPB1 is crucial, because in vivo antisense RNA studies showed that HSPB1 is necessary to inhibit neuronal cell death Citation69. As for other cellular functions of HSPB1, the cell survival‐promoting activity seems to be regulated by phosphorylation Citation69. It has been shown that p38 MAP kinase, which regulates the phosphorylation of HSPB1, plays a fundamental role in the PNS by regulating cell shape and gene expression associated with Schwann cell myelination Citation71.
Increased neuronal cell survival by HSPB1 is not limited to the PNS because several studies indicate its upregulation and protective action also in the central nervous system. In hippocampal and retinal ganglion neurons, Hspb1 is also upregulated after injury Citation72, Citation73. Exogenous HSPB1 protects against neuronal loss in an in vivo model of epilepsy, in a cellular model of Huntington disease, and in an in vitro mammalian cell model of α‐synuclein‐neurotoxicity Citation49, Citation74, Citation75. Furthermore, the neuroprotective effect of increased HSPB1 levels was demonstrated in transgenic HSPB1 mice, which had reduced kainate‐induced seizures and hippocampal cell death Citation76.
It is assumed that loss of constitutive HSPB1 correlates with and promotes neurodegeneration. In the ‘paralyse mouse mutant’, a model of early onset SMA, the expression level of Hspb1 is decreased compared with control mice Citation77. Furthermore, in a cellular model for spinocerebellar ataxia type 3, expressing mutant ataxin‐3, HSPB1 was down‐regulated, suggesting that loss of HSPB1 has a pathologic effect Citation78. HSPB1 expression is also differentially and temporally regulated in mouse models of ALS. Recently, decreased Hspb1 expression was demonstrated several weeks prior to onset of motor neuron death in Cu/Zn superoxide dismutase‐1 (SOD1) mutant mice Citation79. Other studies demonstrated that expression of Hspb1 was gradually upregulated in spinal cords of early and late symptomatic SOD1 mutant mice relative to non‐symptomatic mutant mice Citation20, Citation80.
Until now, the exact mechanism of protection against neuronal cell death is unclear, but the cytoprotective roles of HSPB1 including chaperone‐like activity, anti‐apoptotic activity and preservation of cytoskeletal stability, may all contribute to neuroprotection. The survival‐promoting effect may be associated with the anti‐apoptotic actions of HSPB1. More recently, a direct anti‐apoptotic activity of HSPB1 was shown in a cellular model of Parkinson disease. In this model overexpression of HSPB1 delays the release of cytochrome C, reduces caspase activity and prevents apoptosis Citation81.
Until now there is no direct evidence that HSPB8 is also neuroprotective. Since HSPB8 acts as a cytoprotective component in non‐neuronal cell lines Citation34, it is plausible that it has the ability to protect neurons from cell death. The specific mechanisms underlying this neuroprotection are still to be defined. It will be important to characterize the mechanisms by which HSPB1 and HSPB8 contribute to neuronal survival, because this may lead to the identification of new strategies to prevent neuronal death.
Indeed, compounds inducing HSPs may be useful as a potential treatment for human neurodegenerative diseases. Recently a co‐inducer of HSP expression, arimoclomol, significantly delayed disease progression in a mouse model of ALS, even when administered after the disease onset Citation82. Furthermore, geldanamycin, a naturally occurring anticancer drug, induces HSP expression in different neuronal cell types and suppresses huntingtin protein aggregation in a cellular model of Huntington disease by modulating the expression of HSP40, HSP70 and HSP90 Citation83. Also immunophilin‐ligands, which are used in transplantation to suppress allograft such as FK506, rapidly induce the expression of HSP70 and HSPB1. The administration of FK506 dramatically reduces signs of neuropathy and markedly protects against axonal loss during acrylamide induced neuropathy Citation84. Major drawbacks of these components are the considerable side effects, which limit their application in humans Citation85.
Concluding remarks
The identification of mutations in small HSPs associated with human motor and sensory neuropathies, indicates their pivotal role in the PNS. The diversity of interactions and cytoprotective functions of small HSPs make identifying the underlying pathologic mechanism of these neuromuscular disorders difficult. The mutations could interfere with the chaperone‐like activity or oligomerization property of these small HSPs and may be responsible for the aggregates found in cultured cells expressing the mutant proteins () Citation23, Citation34. Another possibility to explain the induction of neuronal cell death by mutant small HSPs, is their capacity to modulate apoptotic pathways, either directly or indirectly (). Therefore, the HSPB1 mutations observed in patients with inherited neuropathies could interfere with binding of cytochrome C Citation24, Citation50. In this respect, mutations in these small HSPs could also influence the pro‐survival activity of HSPB1 or the pro‐apoptotic activity of HSPB8. Furthermore, the disruption of neurofilament assembly in cells expressing mutant HSPB1 is an indication that this small HSP has a role in the maintenance of cytoskeletal dynamics and axonal transport, which could also be relevant for the pathologic mechanism in these inherited peripheral neuropathies. The length of sensory and motor axons make these cell‐types highly susceptible to defects in axonal transport. The identification of the complete molecular and functional components of small HSPs in motor and sensory neurons will allow understanding why these neurons are specifically affected by mutations in HSPB1 and HSPB8, and will subsequently be useful to develop therapeutic approaches in the future.
If mutations in HSPB1 and HSPB8 result in a loss‐of‐function phenotype, components that upregulate HSP‐expression will be useful for hereditary peripheral neuropathies. However, if mutant small HSPs act as toxic components that induce neuronal cell death, an upregulation of these mutant proteins could make the pathology even worse. The observed mutant phenotype could also be a complex combination of loss‐of‐function and dominant‐negative effects. Therefore, the ideal therapeutic strategy would be to selectively inhibit the expression of the mutant protein and maintain the expression of wild‐type proteins. Hypothetically, since small HSPs are structurally closely related, development of safe therapeutic approaches with the aim to upregulate HSPB1 in patients with mutant HSPB8 and HSPB8 in patients with mutant HSPB1 may improve the neuronal loss observed in distal HMN and CMT2.
Acknowledgements
ID is supported by a PhD fellowship of the Institute for Science and Technology (IWT), and JI is supported by a postdoctoral fellowship of the Fund for Scientific Research (FWO‐Flanders), Belgium.
References
- Gusev N. B., Bogatcheva N. V., Marston S. B. Structure and properties of small heat shock proteins (sHsp) and their interaction with cytoskeleton proteins. Biochemistry (Mosc) 2002; 67: 511–9
- Mounier N., Arrigo A. P. Actin cytoskeleton and small heat shock proteins: how do they interact?. Cell Stress Chaperones 2002; 7: 167–76
- Arrigo A. P. In search of the molecular mechanism by which small stress proteins counteract apoptosis during cellular differentiation. J Cell Biochem 2005; 94: 241–6
- Mehlen P., Mehlen A., Godet J., Arrigo A. P. hsp27 as a switch between differentiation and apoptosis in murine embryonic stem cells. J Biol Chem 1997; 272: 31657–65
- Parcellier A., Gurbuxani S., Schmitt E., Solary E., Garrido C. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophys Res Commun 2003; 304: 505–12
- Wain H. M., Bruford E. A., Lovering R. C., Lush M. J., Wright M. W., Povey S. Guidelines for human gene nomenclature. Genomics 2002; 79: 464–70
- Kappe G., Franck E., Verschuure P., Boelens W. C., Leunissen J. A., De Jong W. W. The human genome encodes 10 alpha‐crystallin‐related small heat shock proteins: HspB1‐10. Cell Stress Chaperones 2003; 8: 53–61
- Franck E., Madsen O., van Rheede T., Ricard G., Huynen M. A., De Jong W. W. Evolutionary diversity of vertebrate small heat shock proteins. J Mol Evol 2004; 59: 792–805
- Mörner C. T. Untersuchung der proteinsubstanzen in den leichtbrechenden medien des auges. Hoppe Seylers Z Physiol Chem 1894; 18: 61–106
- Ingolia T. D., Craig E. A. Four small Drosophila heat shock proteins are related to each other and to mammalian alpha‐crystallin. Proc Natl Acad Sci U S A 1982; 79: 2360–4
- Dubin R. A., Wawrousek E. F., Piatigorsky J. Expression of the murine alpha B‐crystallin gene is not restricted to the lens. Mol Cell Biol 1989; 9: 1083–91
- Hickey E., Brandon S. E., Potter R., Stein G., Stein J., Weber L. A. Sequence and organization of genes encoding the human 27 kDa heat shock protein. Nucleic Acids Res 1986; 14: 4127–45
- Charpentier A. H., Bednarek A. K., Daniel R. L., Hawkins K. A., Laflin K. J., Gaddis S. Effects of estrogen on global gene expression: identification of novel targets of estrogen action. Cancer Res 2000; 60: 5977–83
- Smith C. C., Yu Y. X., Kulka M., Aurelian L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine‐threonine PK and is expressed in melanoma cells. J Biol Chem 2000; 275: 25690–9
- Benndorf R., Sun X. K., Gilmont R. R., Biedermann K. J., Molloy M. P., Goodmurphy C. W. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J Biol Chem 2001; 276: 26753–61
- Kappe G., Verschuure P., Philipsen R. L., Staalduinen A. A., Van de B. P., Boelens W. C. Characterization of two novel human small heat shock proteins: protein kinase‐related HspB8 and testis‐specific HspB9. Biochim Biophys Acta 2001; 1520: 1–6
- Shimura H., Miura‐Shimura Y., Kosik K. S. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 2004; 279: 17957–62
- Renkawek K., Stege G. J., Bosman G. J. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B‐crystallin in Parkinson's disease. Neuroreport 1999; 10: 2273–6
- Head M. W., Corbin E., Goldman J. E. Overexpression and abnormal modification of the stress proteins alpha B‐crystallin and HSP27 in Alexander disease. Am J Pathol 1993; 143: 1743–53
- Vleminckx V., Van Damme P., Goffin K., Delye H., Van Den B. L., Robberecht W. Upregulation of HSP27 in a transgenic model of ALS. J Neuropathol Exp Neurol 2002; 61: 968–74
- Litt M., Kramer P., LaMorticella D. M., Murphey W., Lovrien E. W., Weleber R. G. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet 1998; 7: 471–4
- Vicart P., Caron A., Guicheney P., Li Z., Prevost M. C., Faure A. A missense mutation in the alphaB‐crystallin chaperone gene causes a desmin‐related myopathy. Nat Genet 1998; 20: 92–5
- Irobi J., Van Impe K., Seeman P., Jordanova A., Dierick I., Verpoorten N. Hot‐spot residue in small heat‐shock protein 22 causes distal motor neuropathy. Nat Genet 2004; 36: 597–601
- Evgrafov O. V., Mersiyanova I. V., Irobi J., Van den Bosch L., Dierick I., Schagina O. Mutant small heat‐shock protein 27 causes axonal Charcot‐Marie‐Tooth disease and distal hereditary motor neuropathy. Nat Genet 2004; 36: 602–6
- Tang B. S., Zhao G. H., Luo W., Xia K., Cai F., Pan Q. Small heat‐shock protein 22 mutated in autosomal dominant Charcot‐Marie‐Tooth disease type 2L. Hum Genet 2005; 116: 222–4
- Emery A. E. H. Review: The nosology of the spinal muscular atrophies. J Med Genet 1971; 8: 481–95
- Harding A. E., Thomas P. K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980; 103: 259–80
- Irobi J., De Jonghe P., Timmerman V. Molecular genetics of distal hereditary motor neuropathies. Hum Mol Genet 2004; 13: 195–202
- Shy M. E. Charcot‐Marie‐Tooth disease: an update. Curr Opin Neurol 2004; 17: 579–85
- Tang B. S., Luo W., Xia K., Xiao J. F., Jiang H., Shen L. A new locus for autosomal dominant Charcot‐Marie‐Tooth disease type 2 (CMT2L) maps to chromosome 12q24. Hum Genet 2004; 114: 527–33
- Bera S., Thampi P., Cho W. J., Abraham E. C. A positive charge preservation at position 116 of alpha A‐crystallin is critical for its structural and functional integrity. Biochemistry 2002; 41: 12421–6
- Andley U. P., Patel H. C., Xi J. H. The R116C mutation in alpha A‐crystallin diminishes its protective ability against stress‐induced lens epithelial cell apoptosis. J Biol Chem 2002; 277: 10178–86
- Fu L., Liang J. J. Alteration of protein‐protein interactions of congenital cataract crystallin mutants. Invest Ophthalmol Vis Sci 2003; 44: 1155–9
- Zobel A. T. C., Loranger A., Marceau N., Theriault J. R., Lambert H., Landry J. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy‐causing R120G alpha B‐crystallin mutant. Hum Mol Genet 2003; 12: 1609–20
- Muchowski P. J., Wu G. J., Liang J. J., Adman E. T., Clark J. I. Site‐directed mutations within the core "alpha‐crystallin" domain of the small heat‐shock protein, human alphaB‐crystallin, decrease molecular chaperone functions. J Mol Biol 1999; 289: 397–411
- Pasta S. Y., Raman B., Ramakrishna T., Rao C. The IXI/V motif in the C‐terminal extension of alpha‐crystallins: alternative interactions and oligomeric assemblies. Mol Vis 2004; 10: 655–62
- Studer S., Obrist M., Lentze N., Narberhaus F. A critical motif for oligomerization and chaperone activity of bacterial alpha‐heat shock proteins. Eur J Biochem 2002; 269: 3578–86
- Pasta S. Y., Raman B., Ramakrishna T., Rao C. Role of the conserved SRLFDQFFG region of alpha‐crystallin, a small heat shock protein. Effect on oligomeric size, subunit exchange, and chaperone‐like activity. J Biol Chem 2003; 278: 51159–66
- Diaz‐Latoud C., Buache E., Javouhey E., Arrigo A. P. Substitution of the unique cysteine residue of murine hsp25 interferes with the protective activity of this stress protein through inhibition of dimer formation. Antioxid Redox Signal 2005; 7: 436–45
- Chowdary T. K., Raman B., Ramakrishna T., Rao C. M. Mammalian Hsp22 is a heat‐inducible small heat‐shock protein with chaperone‐like activity. Biochem J 2004; 381: 379–87
- Kim M. V., Seit‐Nebi A. S., Marston S. B., Gusev N. B. Some properties of human small heat shock protein Hsp22 (H11 or HspB8). Biochem Biophys Res Commun 2004; 315: 796–801
- Sun X., Fontaine J. M., Rest J. S., Shelden E. A., Welsh M. J., Benndorf R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J Biol Chem 2004; 279: 2394–402
- Giese K. C., Vierling E. Changes in oligomerization are essential for the chaperone activity of a small heat shock protein in vivo and in vitro. J Biol Chem 2002; 277: 46310–8
- Rogalla T., Ehrnsperger M., Preville X., Kotlyarov A., Lutsch G., Ducasse C. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem 1999; 274: 18947–56
- Haslbeck M., Buchner J. Chaperone function of sHsps. Prog Mol Subcell Biol 2002; 28: 37–59
- Jakob U., Gaestel M., Engel K., Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem 1993; 268: 1517–20
- Theriault J. R., Lambert H., Chavez‐Zobel A. T., Charest G., Lavigne P., Landry J. Essential role of the NH2‐terminal WD/EPF motif in the phosphorylation‐activated protective function of mammalian Hsp27. J Biol Chem 2004; 279: 23463–71
- Lindner R. A., Carver J. A., Ehrnsperger M., Buchner J., Esposito G., Behlke J. Mouse Hsp25, a small shock protein. The role of its C‐terminal extension in oligomerization and chaperone action. Eur J Biochem 2000; 267: 1923–32
- Wyttenbach A., Sauvageot O., Carmichael J., Diaz‐Latoud C., Arrigo A. P., Rubinsztein D. C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet 2002; 11: 1137–51
- Bruey J. M., Ducasse C., Bonniaud P., Ravagnan L., Susin S. A., Diaz‐Latoud C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2000; 2: 645–52
- Pandey P., Farber R., Nakazawa A., Kumar S., Bharti A., Nalin C. Hsp27 functions as a negative regulator of cytochrome c‐dependent activation of procaspase‐3. Oncogene 2000; 19: 1975–81
- Charette S. J., Lavoie J. N., Lambert H., Landry J. Inhibition of Daxx‐mediated apoptosis by heat shock protein 27. Mol Cell Biol 2000; 20: 7602–12
- Parcellier A., Schmitt E., Gurbuxani S., Seigneurin‐Berny D., Pance A., Chantome A. HSP27 is a ubiquitin‐binding protein involved in I‐kappaBalpha proteasomal degradation. Mol Cell Biol 2003; 23: 5790–802
- Gober M. D., Smith C. C., Ueda K., Toretsky J. A., Aurelian L. Forced expression of the h11 heat shock protein can be regulated by DNA methylation and trigger apoptosis in human cells. J Biol Chem 2003; 278: 37600–9
- Hase M., Depre C., Vatner S. F., Sadoshima J. H11 has dose‐dependent and dual hypertrophic and proapoptotic functions in cardiac myocytes. Biochem J 2005; 388: 475–83
- Murashov A. K., Ul H., I., Hill C., Park E., Smith M., Wang X. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res Mol Brain Res 2001; 93: 199–208
- Depre C., Hase M., Gaussin V., Zajac A., Wang L., Hittinger L. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ Res 2002; 91: 1007–14
- Benndorf R., Hayess K., Ryazantsev S., Wieske M., Behlke J., Lutsch G. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization‐inhibiting activity. J Biol Chem 1994; 269: 20780–4
- Guay J., Lambert H., Gingras‐Breton G., Lavoie J. N., Huot J., Landry J. Regulation of actin filament dynamics by p38 map kinase‐mediated phosphorylation of heat shock protein 27. J Cell Sci 1997; 110(Pt 3): 357–68
- Wieske M., Benndorf R., Behlke J., Dolling R., Grelle G., Bielka H. Defined sequence segments of the small heat shock proteins HSP25 and alphaB‐crystallin inhibit actin polymerization. Eur J Biochem 2001; 268: 2083–90
- Perng M. D., Cairns L., van den IJssel P., Prescott A., Hutcheson A. M., Quinlan R. A. Intermediate filament interactions can be altered by HSP27 and alpha B‐crystallin. J Cell Sci 1999; 112: 2099–112
- Mersiyanova I. V., Perepelov A. V., Polyakov A. V., Sitnikov V. F., Dadali E. L., Oparin R. B. A new variant of Charcot‐Marie‐Tooth disease type 2 (CMT2E) is probably the result of a mutation in the neurofilament light gene. Am J Hum Genet 2000; 67: 37–46
- Gros‐Louis F., Lariviere R., Gowing G., Laurent S., Camu W., Bouchard J. P. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem 2004; 279: 45951–6
- Züchner S., Noureddine M., Kennerson M., Verhoeven K., Claeys K., De Jonghe P. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot‐Marie‐Tooth disease. Nat Genet 2005; 37: 289–94
- Puls I., Jonnakuty C., LaMonte B. H., Holzbaur E. L., Tokito M., Mann E. Mutant dynactin in motor neuron disease. Nat Genet 2003; 33: 455–6
- Costigan M., Mannion R. J., Kendall G., Lewis S. E., Campagna J. A., Coggeshall R. E. Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J Neurosci 1998; 18: 5891–900
- Hirata K., He J., Hirakawa Y., Liu W., Wang S., Kawabuchi M. HSP27 is markedly induced in Schwann cell columns and associated regenerating axons. Glia 2003; 42: 1–11
- Lewis S. E., Mannion R. J., White F. A., Coggeshall R. E., Beggs S., Costigan M. A role for HSP27 in sensory neuron survival. J Neurosci 1999; 19: 8945–53
- Benn S. C., Perrelet D., Kato A. C., Scholz J., Decosterd I., Mannion R. J. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 2002; 36: 45–56
- Wagstaff M. J., Collaco‐Moraes Y., Smith J., de Belleroche J. S., Coffin R. S., Latchman D. S. Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus‐based vector. J Biol Chem 1999; 274: 5061–9
- Fragoso G., Robertson J., Athlan E., Tam E., Almazan G., Mushynski W. E. Inhibition of p38 mitogen‐activated protein kinase interferes with cell shape changes and gene expression associated with Schwann cell myelination. Exp Neurol 2003; 183: 34–46
- Krueger‐Naug A. M., Emsley J. G., Myers T. L., Currie R. W., Clarke D. B. Injury to retinal ganglion cells induces expression of the small heat shock protein Hsp27 in the rat visual system. Neuroscience 2002; 110: 653–65
- Valentim L. M., Geyer A. B., Tavares A., Cimarosti H., Worm P. V., Rodnight R. Effects of global cerebral ischemia and preconditioning on heat shock protein 27 immunocontent and phosphorylation in rat hippocampus. Neuroscience 2001; 107: 43–9
- Kalwy S. A., Akbar M. T., Coffin R. S., de Belleroche J., Latchman D. S. Heat shock protein 27 delivered via a herpes simplex virus vector can protect neurons of the hippocampus against kainic‐acid‐induced cell loss. Brain Res Mol Brain Res 2003; 111: 91–103
- Zourlidou A., Payne S., Latchman D. S. HSP27 but not HSP70 has a potent protective effect against alpha‐synuclein‐induced cell death in mammalian neuronal cells. J Neurochem 2004; 88: 1439–48
- Akbar T. M., Lundberg A. M., Liu K., Vidyadaran S., Wells K. E., Dolatshad H. The neuroprotective effects of heat shock protein 27 overexpression in transgenic animals against kainate‐induced seizures and hippocampal cell death. J Biol Chem 2003; 278: 19956–65
- Pieri I., Cifuentes‐Diaz C., Oudinet J. P., Blondet B., Rieger F., Gonin S. Modulation of HSP25 expression during anterior horn motor neuron degeneration in the paralyse mouse mutant. J Neurosci Res 2001; 65: 247–53
- Wen F. C., Li Y. H., Tsai H. F., Lin C. H., Li C., Liu C. S. Down‐regulation of heat shock protein 27 in neuronal cells and non‐neuronal cells expressing mutant ataxin‐3. FEBS Lett 2003; 546: 307–14
- Maatkamp A., Vlug A., Haasdijk E., Troost D., French P. J., Jaarsma D. Decrease of Hsp25 protein expression precedes degeneration of motoneurons in ALS‐SOD1 mice. Eur J Neurosci 2004; 20: 14–28
- Batulan Z., Shinder G. A., Minotti S., He B. P., Doroudchi M. M., Nalbantoglu J. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 2003; 23: 5789–98
- Gorman A. M., Szegezdi E., Quigney D. J., Samali A. Hsp27 inhibits 6‐hydroxydopamine‐induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun 2005; 327: 801–10
- Kieran D., Kalmar B., Dick J. R., Riddoch‐Contreras J., Burnstock G., Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 2004; 10: 402–5
- Sittler A., Lurz R., Lueder G., Priller J., Lehrach H., Hayer‐Hartl M. K. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet 2001; 10: 1307–15
- Gold B. G., Voda J., Yu X., Gordon H. The immunosuppressant FK506 elicits a neuronal heat shock response and protects against acrylamide neuropathy. Exp Neurol 2004; 187: 160–70
- Klettner A. The induction of heat shock proteins as a potential strategy to treat neurodegenerative disorders. Drug News Perspect 2004; 17: 299–306