Abstract
BACKGROUND. Inhibition of tumour cell proliferation, invasion and metastasis by chemically modified tetracyclines has been ascribed to inhibition of matrix metalloproteinase (MMP) activity.
METHODS. Exposure of the human breast carcinoma cell line MDA‐MB‐231 or its MMP‐9‐overproducing transfected clone (E‐10) to 6‐demethyl, 6‐deoxy, 4‐de [dimethylamino]‐tetracycline (CMT‐3), a chemically modified non‐antimicrobial tetracycline followed by analysis using gelatinase activity assay, zymography, degradation of radiolabelled extracellular matrix (ECM), Western blotting, TNF‐α ELISA and cell viability assays.
RESULTS. CMT‐3 treatment results in diminution in extracellular MMP‐9 protein levels as well as inhibition of gelatinase activity. This prevents cell‐mediated ECM degradation without inducing general cytostasis or cytotoxicity. Culturing E‐10 cells in 10 or 20 µM CMT‐3 diminished secreted MMP‐9 levels by 45% or 60%, respectively, but did not affect levels of most other secreted proteins, including tissue inhibitor of Metalloproteinases (TIMP‐1). ECM degradation by E‐10 cells or their conditioned medium was inhibited by ∼ 20%–30% in the presence of 20 µM CMT‐3, reflecting inhibition of MMP‐9 activity in addition to diminution of released MMP‐9 levels. TNF‐α levels were also diminished in E‐10 conditioned medium in the presence of CMT‐3, but cell viability, measured by MTS reduction and cytosolic LDH retention, was unaffected.
CONCLUSIONS. It is proposed that the reduction in ECM‐degradative activity reflects diminished levels of expression as well as inhibition of enzymatic activity of MMPs released by cells in the presence of CMT‐3. These multiple effects of CMT‐3 may offer promise for use in suppressing tumour invasion, and if used in conjunction with other chemotherapy agents, may lead to more successful treatment of cancer.
| Abbreviations | ||
| DMEM | = | Dulbecco's modified eagle medium |
| MEM | = | minimum essential medium |
| ECM | = | extracellular matrix |
| MMP(s) | = | matrix metalloproteinase(s) |
| TIMP | = | tissue inhibitor of matrix metalloproteinase |
| SFM | = | serum free medium |
| TC | = | tetracycline |
| CMT | = | chemically modified tetracycline |
| FBS | = | foetal bovine serum |
| PBS | = | phosphate‐buffered saline |
| PMSF | = | phenylmethanesulfonyl fluoride |
| APMA | = | p‐aminophenylmercuric acetate |
| mAb | = | monoclonal antibody |
| MTS | = | 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt |
| LDH | = | lactate dehydrogenase |
| HRP | = | horse radish peroxide |
| TMB | = | tetramethylbenzidine |
| NADH | = | nicotinamide adenine dinucleotide‐hydrogen |
| VEGF | = | vascular endothelial growth factor. |
Introduction
A major source of the morbidity and mortality of breast cancer is the highly invasive and metastatic potential of these tumours. In order for tumour cells to metastasize, they must pass through the extracellular matrix (ECM) of the interstitial stroma as well as the basement membrane underlying the vascular endothelium. Mechanisms of invasion and metastasis by various types of breast tumours involve proteolytic activity derived from the tumour and the surrounding stroma, either separately or jointly Citation1, Citation2. Considerable interest has recently been directed to characterization of the roles of the class of proteinases known as matrix metalloproteinases (MMPs) in invasion and metastasis of breast tumours Citation1. The MMPs share several common features, including dependence on zinc for catalytic activity, secretion as zymogens, activation by proteinases, oxidants or organomercurials, digestion of components of ECM, and inhibition by tissue inhibitors of MMPs (TIMPs). MMP expression can be regulated, for example, by growth factors and cytokines such as interleukins‐1 and ‐4, transforming growth factors‐α and ‐β, and tumour necrosis factor‐α Citation3–7. In view of their ability to degrade components of the basement membrane as well as interstitial stroma, the extracellular and membrane‐bound MMPs have been implicated in tumour cell invasion and metastasis. On the other hand, recent studies using MMP‐8 knock‐out versus wild‐type mice have shown that certain MMPs, i.e. MMP‐8, can unexpectedly protect against the spread of experimental cancer and inflammation by processing of anti‐inflammatory mediators that can contribute to the host's protective anti‐tumour anti‐inflammatory systems Citation8. In this regard it is now becoming apparent that a complete inhibition of MMPs in general may not be desired Citation8, Citation9, but instead only the reduction of pathologically excessive MMPs without interfering with their defensive action would be optimal for therapeutic purposes Citation8, Citation10. Various genetically distinct but structurally related MMPs have been detected in human cancer tissues as well as in serum and urine of cancer patients Citation11, Citation12. This evidence has prompted the development of MMP inhibitors to be used in anti‐invasive or anti‐metastatic therapy for management of cancers as well as for a variety of inflammatory diseases Citation10.
The discovery of the MMP‐inhibitory activity of tetracyclines (TCs) has led to the creation of a family of chemically modified tetracyclines (CMTs) in which MMP‐inhibitory activity has been augmented but antibiotic activity has been eliminated Citation10. Even TCs which retained antimicrobial activity were found to inhibit melanoma cell collagenase activity as well as cell invasion and metastasis Citation11. TCs have also been shown to inhibit the 72‐kD gelatinase activity from purified human lung cancer cells Citation12. More recently, newly designed CMTs such as 6‐demethyl‐6‐deoxy‐4‐de(dimethylamino)tetracycline (CMT‐3) were found to be more potent inhibitors of MMP activity, cell proliferation, invasion, tumour growth, and metastasis than any of the antimicrobially active TCs Citation13, Citation14. In a number of prostate tumour cell lines, CMT‐3 induced arrest of cell cycle progression, loss of mitochondrial membrane potential, and apoptosis in addition to diminution in levels of MMPs and TIMPs Citation14.
In an effort to dissect the apparent multiplicity of effects of CMT‐3 on tumour cells, we have utilized the human breast carcinoma cell line MDA‐MB‐231 as well as an MMP‐9‐overexpressing stably transfected clone of this line (the E‐10 subclone developed by Ramos‐DeSimone et al. Citation15), to study the effect of CMT‐3 on breast tumour‐mediated ECM degradation and invasion. We quantitated the degradative activity of these cells as a measure of their invasive potential by employing a complete interstitial ECM that had been biosynthesized by cultured stromal cells in the presence of radiolabelled precursors. The labels become incorporated into the matrix components: 37% to fibrillar collagens, 11% to elastin, and 52% to trypsin‐sensitive proteoglycans and adhesion glycoproteins Citation16. Degradative activity is measured by solubilization of the radiolabelled ECM. We have demonstrated previously that MMPs participate in the ECM degradation process mediated by tumour cells Citation17. Here we describe studies used to evaluate the dose‐dependent inhibition of tumour cell gelatinolytic and invasive activity by CMT‐3 as well as discuss the possible mechanisms of the CMT‐3 inhibition.
Key messages
CMT‐3 diminishes extracellular MMP‐9 protein levels and activity.
CMT‐3 diminished invasion of breast carcinoma‐derived E‐10 cells into extracellular matrix.
As CMT‐3 does not lead to diarrhoea or microbial resistance, it might find future use as an adjunct therapy in the treatment of breast cancer.
Material and methods
Chemicals
Cell culture reagents were purchased from Hyclone, Inc. (Logan, UT). Phenylmethanesulfonyl fluoride (PMSF), and p‐aminophenylmercuric acetate (APMA) were from Sigma (St. Louis, MO). Radiolabels were from ICN biomedical Inc. (Costa Mesa, CA). All monoclonal antibodies (mAbs) against individual MMPs were purchased from Oncogene Research Products (Cambridge, MA). SDS‐PAGE gels and gelatine zymogram gels were from Novel Experimental Technology (San Diego, CA). Secondary antibodies for Western blotting were purchased from Kirkegaard & Perry Laboratories, Inc. (Gaithersburg, MD). ECL Western blotting detection reagents were from Amersham Life Science Inc. (Cleveland, OH). CMTs were kindly provided by Collagenex Pharmaceuticals, Inc. (Newtown, PA).
Cell Culture
The breast carcinoma‐derived line MDA‐MB‐231 was obtained from the American Type Culture Commission. The E‐10 stable transfected subclone of MDA‐MB‐231 was kindly provided by Dr. James Quigley (Scripps Institute, La Jolla, CA). Cells were cultured in 75‐cm2 T‐flasks in Dulbecco's modified eagle medium (DMEM) supplemented with 10% heat‐inactivated FBS‐containing 100 unit/mL penicillin and 100 µg/mL streptomycin. Cells were grown to 80%–90% confluence at which time the 10% FBS‐containing medium was replaced with serum‐free medium (SFM) to prevent serum‐inhibition and/or activation of MMP activity. Conditioned medium was collected after 48 hours and centrifuged to remove cell debris. Unless otherwise indicated, this conditioned medium was collected from cells grown on plastic. In some experiments, cells were maintained for 48 hours in SFM containing CMT‐3. For experiments studying the total protein synthesis in E‐10 cells, cells were grown to 80%–90% confluence at which time the 10% FBS‐containing medium was replaced with SFM containing [35S]‐methionine/cysteine (TransLabel). Conditioned medium was collected after 48 hours and examined by SDS‐PAGE fluorography. All experiments were performed in triplicate at least twice and analyzed by gelatine zymography, Western blotting or gelatinase assay.
Preparation of wells coated with radiolabelled ECM
R22 rat heart smooth muscle cells were cultured in minimum essential medium (MEM) supplemented with FBS, tryptose phosphate broth, and cefotaxime. The R22 cells were plated onto multiwell tissue culture plates at an initial density of 2.5×104 cells/cm2 and were maintained at 37°C in 5% CO2. At confluence, the medium was supplemented with [3H]‐proline which was incorporated into a complete interstitial ECM elaborated by the cells Citation17. Every four days 50 µg/mL ascorbic acid was added to ensure maximal yield of a collagen‐rich, insoluble ECM. After culturing for at least one week in radiolabelled medium, cells were lysed by addition of 25 mM NH4OH without disrupting the ECM. The wells were washed three times with sterile H2O and once in PBS containing 0.02% NaN3. Excess PBS was then removed and plates were stored at 4°C until use. Before use, the ECM was rehydrated by rinsing three times with sterile buffer.
Degradation of radiolabelled ECM
Cells at a density of 2.8×105/mL or conditioned medium were applied to R22 ECM‐coated wells of microplates and incubated for 2 days at 37oC in 5% CO2. Different reagents were added to separate wells. After two‐day incubation, supernatants were collected and the remaining undegraded ECM in each well was solubilized by overnight incubation with 2 M NaOH. The radioactivity in the supernatants and in the NaOH was determined in a Liljeholmens Stearinfabrik, Kema och Stockholms Bryggerier (LKB) liquid scintillation counter. Experiments were performed in triplicate wells.
Gelatine zymography
SDS‐PAGE gels containing copolymerized gelatine at a final concentration of 1 mg/mL were prepared. After electrophoresis, the gels were washed in 2.5% Triton X‐100 and incubated at 37°C overnight in calcium assay buffer (40 mM Tris, 200 mM NaCl, 10 mM CaCl2, pH 7.5). After incubation, the gels were stained with Coomassie Brilliant Blue R‐250. Clear zones of lysis against a blue background indicated gelatinolytic activity.
Immunoblotting
Samples were separated by 8% SDS‐PAGE and transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies overnight and subsequently with alkaline phosphatase (AP)‐conjugated secondary antibodies for 2 hours. After washing, the immunoblots were incubated with chemiluminescent substrate and exposed to X‐ray film. The band densities were scanned by an LKB Ultroscan XL laser densitometer.
Gelatinase assay
Gelatinolytic activity of the conditioned medium was assayed using heat denatured [3H]‐labelled collagen Citation13. Samples of E‐10 conditioned medium were incubated with [3H]‐gelatine substrate at 37°C overnight, followed by treatment with 45% trichloroacetic acid (TCA) at 4°C for 30 minutes and centrifugation to precipitate the undigested substrate. The radioactivity in the supernatants was determined in an LKB liquid scintillation counter.
TNF‐α ELISA assay
TNF‐α ELISA kits were purchased from Endogen, Inc. (Rockford, IL). 50 µL of the reconstituted standards or samples were plated onto wells of plates coated with anti‐human TNF‐α capture antibody followed by an incubation with 50 µL of a biotinylated detection antibody reagent at room temperature for two hours. At the end of incubation, the plate was washed three times and 100 µl of streptavidin‐horse radish peroxide (HRP) solution was added to each well and incubated for 30 minutes at room temperature. Following another three washes, 100 µl of tetramethylbenzidine (TMB) substrate solution was added to each well and the coloured product was allowed to develop at room temperature in the dark. After 30 minutes, 100 µL of stop solution was added and the absorbance of the samples was measured at 450 nm by an ELISA reader (Packard SpectraCountTM , Palo Alto, CA). Experiments were performed in triplicate.
Cell viability assays
Cell viability was assayed by reduction of the tetrazolium salt 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt (MTS) to formazan (Promega, Madison, WI). E‐10 cells were plated onto ECM‐coated wells in the presence of concentrations of CMT‐3 ranging from 5 µM to 100 µM and cultured for 48 hours. At that time, 20 µL of a MTS/phenazine methosulfate solution per 0.1 mL medium was added to each well, and after 2 hours of incubation, the levels of solubilized formazan were quantitated by measurement of absorbance at 490 nm. To confirm that loss of MTS reductase activity was due to cytotoxicity of CMT‐3, release of the cytosolic enzyme lactate dehydrogenase (LDH) into the supernatant medium was assayed (Sigma, St. Louis, MO). Sodium pyruvate containing 1 mg/mL nicotinamide adenine dinucleotide‐hydrogen (NADH) was added to 0.1 mL medium at 37°C for 0.5 hour; 1.0 mL 2,4‐dinitrophenylhydrazine reagent was then added at room temperature for 20 minutes. 10 mL 0.4N NaOH was finally added and the level of LDH which had been liberated into the supernatant was determined by measurement of the decrement in absorbance at 490 nm as pyruvate was converted to lactate. Experiments were performed in triplicate wells.
Results
Effect of CMT‐3 on ECM degradation by E‐10 cells and conditioned medium
When E‐10 conditioned medium was incubated without any CMT‐3 added for two days on [3H]‐proline labelled ECM, ∼10% of the total matrix‐associated proline counts were solubilized, consistent with previously observed degradative activity of tumour cell line CM preparations Citation17. This matrix‐dependent solubilization was inhibited by CMT‐3, e.g. by 25% when 10 µM CMT‐3 was present, and by 30% in the presence of 20 µM CMT‐3 (). These results were confirmed by gelatine zymography (). E‐10 cells maintained for two days in SFM on same ECM degraded the ECM to an even greater extent. About 35 2% of the proline‐labeled ECM was solubilized by cells in the absence of CMT‐3. This cell‐mediated degradation was decreased by 15% in the presence of 10 µM CMT‐3 and by 20% when cells were cultured in SFM in the presence of 20 µM CMT‐3 (data not shown). E‐10 is a subclone isolated from a transfection of the MDA‐MB 231 breast cancer cell line with multiple copies of the MMP‐9 gene Citation15. When its parental cell line, MDA‐MB 231 cells, were maintained for two days in SFM on ECM, MDA‐MB 231 cells only degraded about 6% of the ECM whereas E‐10 cells degraded 34±2% of the ECM ().
Figure 1 Effect of CMT‐3 on E‐10 conditioned medium as assessed by gelatinase activity assay (upper panel) and zymography (lower panel). E‐10 conditioned medium was plated on a [3H]‐proline labelled ECM plate. Different concentrations of CMT‐3 were added to selected wells at the start of the 2‐day incubation. Supernatants were removed and assayed for radioactivity. The residual radioactivity in each well was then solubilized with NaOH and measured. Each value represents triplicates (mean±standard deviation). In the lower panel E‐10 conditioned medium was instead analyzed by zymography.
![Figure 1 Effect of CMT‐3 on E‐10 conditioned medium as assessed by gelatinase activity assay (upper panel) and zymography (lower panel). E‐10 conditioned medium was plated on a [3H]‐proline labelled ECM plate. Different concentrations of CMT‐3 were added to selected wells at the start of the 2‐day incubation. Supernatants were removed and assayed for radioactivity. The residual radioactivity in each well was then solubilized with NaOH and measured. Each value represents triplicates (mean±standard deviation). In the lower panel E‐10 conditioned medium was instead analyzed by zymography.](/cms/asset/4d661fcb-bcd5-4acd-9562-6b4945af2a95/iann_a_130021_f0001_b.jpg)
Figure 2 ECM degradative activity mediated by MDA‐MB231 versus E‐10 cells. MDA‐MB 231 or E‐10 cells at a density of 2.8×105 cells/cm2 were plated on a [3H]‐proline labelled ECM plate. Supernatants were removed after 2‐day incubation and assayed for radioactivity. The residual radioactivity in each well was then solubilized with NaOH and determined.
![Figure 2 ECM degradative activity mediated by MDA‐MB231 versus E‐10 cells. MDA‐MB 231 or E‐10 cells at a density of 2.8×105 cells/cm2 were plated on a [3H]‐proline labelled ECM plate. Supernatants were removed after 2‐day incubation and assayed for radioactivity. The residual radioactivity in each well was then solubilized with NaOH and determined.](/cms/asset/04ab45b3-61ee-4f58-b095-bd0b06d3b47a/iann_a_130021_f0002_b.jpg)
Effect of CMT‐3 on gelatinase activity
The inhibitory effect of CMT‐3 on the total gelatinolytic activity in the conditioned medium from E‐10 cells was evaluated by an assay of degradation of gelatine into acid soluble products. E‐10 conditioned medium was incubated with 1 mM APMA at 37°C for one hour first to convert latent MMPs to their active forms before the gelatine substrate was added, along with different concentrations of CMT‐3, and the samples incubated overnight. The total gelatinolytic activity in E‐10 conditioned medium alone with any APMA converted 3±1% and with APMA 55±2% of the gelatine to TCA‐soluble products. In the presence of 10 µM CMT‐3, 46±1% of the gelatine was cleaved to TCA‐soluble products, and in the presence of 20 µM CMT, only 25±1% of the gelatine was degraded. This demonstrated that CMT‐3 exhibited the ability to inhibit the gelatinase activity released into the surrounding medium by this breast tumour cell line. Noteworthy, the inhibition of the gelatinase activity by 10 µM and 20 µM CMT‐3 was significant but not complete.
Effect of CMT‐3 on extracellular MMP‐9 levels
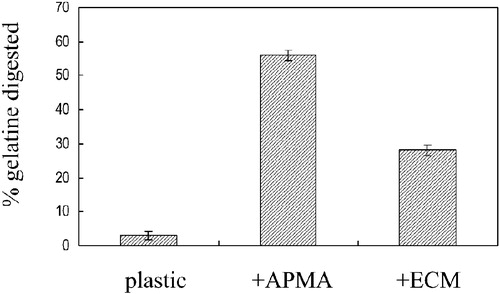
The extracellular levels of MMP‐9 in the conditioned medium collected from cells maintained in the presence of 10 µM and 20 µM concentrations of CMT‐3 were initially analyzed by Western blotting and quantitated by laser densitometry. When E‐10 cells were cultured in the presence of 10 µM CMT‐3, the levels of MMP‐9 in the conditioned medium were reduced by ∼45%. When cells were cultured in the presence of 20 µM CMT‐3 for 48 hours, levels of MMP‐9 in E‐10 conditioned medium were reduced by ∼60% (). After separation of the proteins in the concentrated conditioned medium by SDS‐PAGE, the gels were soaked in renaturation buffer in the absence of any CMTs for zymogram development. Diminished MMP‐9 levels were observed in the conditioned medium collected from cells cultured in the presence of CMT‐3 (). When non‐transfected MDA‐MB‐231 cells were cultured in the absence or presence of CMT‐3, the levels of MMP‐9 visualized by gelatine zymography were uniformly lower than those present in the conditioned medium collected from the transfected E‐10 cells, but a similar dose‐dependent diminution in MMP‐9 levels could be seen in the presence of CMT‐3 (data not shown). Gelatine zymographies also revealed that in E‐10 subclone () or the MDA‐MB‐231 cell (data not shown) conditioned medium not only the latent MMP‐9, but also a lower molecular weight band migrating in the vicinity of known activated forms of the MMP‐9 could be detected. An assay of gelatinolytic activity of E‐10 conditioned medium which had not been treated with APMA to convert latent MMPs into their active forms confirmed that proteolysis by endogenous active MMP‐9 was markedly increased when the cells had been maintained on the R‐22 ECM instead of plastic (). When conditioned medium collected from E‐10 cells cultured on plastic was incubated with [3H]‐gelatine substrate, only 3% of gelatine was digested, but when the conditioned medium was collected from cells cultured on the R22 ECM, the extent of digestion was 28%. The increase in gelatinolytic activity of MMP‐9 in the conditioned medium from E‐10 cells cultured on ECM was closely correlated with the generation of lower molecular weight, putatively active species observed on gelatine zymograms. It may be that the more extensive ECM degradation mediated by E‐10 cells compared to their cell‐free conditioned medium as described above could also reflect the production of this active species. It should be noted that whereas interaction of E‐10 cells with ECM enhanced endogenous activation of MMP‐9, CMT‐3 appeared to reduce the levels of both the latent pro‐MMP‐9 and lower molecular weight activated MMP‐9 in a dose‐response fashion ().
Figure 3 Effect of CMT‐3 on extracellular MMP‐9 levels in E‐10 cells (Western blot analysis). Cells were cultured in serum‐free medium in the absence or presence of CMT‐3 for 48 hours. Conditioned medium was collected and centrifuged to remove cell debris. 2 µL aliquots were analyzed by Western blotting, using an anti‐MMP‐9 mAb. The location of a 92 kDa standard is indicated by the arrow.

Figure 4 Effect of CMT‐3 on extracellular MMP‐9 in E‐10 cells analyzed by gelatinase activity assay. Cells were plated on ECM‐coated wells and incubated for 48 hours before supernatants were collected and analyzed by gelatine activity assay without and with APMA.
Effect of CMT‐3 on TIMP‐1 production
Production of MMPs by tumour cells is often accompanied by production of the endogenous MMP inhibitors, tissue inhibitors of metalloproteinases (TIMPs). TIMP‐1 can inhibit the catalytic activity of MMP‐9 as well as other MMPs through interactions with its amino‐terminal domain, while it also specifically inhibits the conversion of pro‐MMP‐9 to its active form through interactions with its carboxy‐terminal domain. Levels of TIMP‐1 in the concentrated conditioned medium from E‐10 cells cultured in the presence of CMT‐3 were measured by densitometry of Western blots, using TIMP‐1 specific mAbs and using the same procedure as that described above for studies on levels of MMP‐9 (). CMT‐3 was found to have little to no effect on levels of TIMP‐1 in E‐10 conditioned medium.
Figure 5 Effect of CMT‐3 on extracellular TIMP‐1 levels in E‐10 cells. Cells were cultured in serum‐free medium containing different concentrations of CMT‐3 for 48 hours. Conditioned medium was collected and centrifuged to remove cell debris. Samples were analyzed by Western blotting, using an anti‐TIMP‐1 mAb. The location of a TIMP‐1 standard is indicated by the arrow.

Effect of CMT‐3 on TNF‐α production
MMP expression has been shown to be responsive to cytokines. Of particular interest, TNF‐α has been reported to trigger an increase in MMP‐9 levels Citation18. Thus, the effect of CMT‐3 on extracellular TNF‐α levels was investigated to search for the possible mechanisms of CMT‐3 inhibition of MMP‐9 levels. E‐10 cells were found to release 78.32 pg/mL of TNF‐α to the surrounding environment. In the presence of 10 µM CMT‐3 the TNF‐α levels were reduced to 39.65 pg/mL, a diminution of 49%. In the presence of 20 µM CMT‐3 the levels were reduced to 24.23 pg/mL, a diminution of 69%.
Effect of CMT‐3 on total protein synthesis
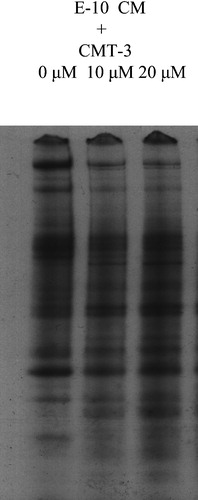
To investigate if CMT‐3 has a general effect on protein synthesis, E‐10 cells were cultured in [35S]‐methionine/cysteine (TransLabel)‐containing SFM in the presence of 10 µM and 20 µM CMT‐3. Conditioned medium was collected and examined by SDS‐PAGE fluorography (). The intensities of the multiple bands on the fluorogram were not significantly different in the conditioned medium from CMT‐treated and control cells. This close similarity in patterns and levels of secreted proteins refutes the possibility that CMT‐3 inhibits total protein synthesis in E‐10 cells; instead, it affected levels of a limited number of secreted proteins selectively.
Figure 6 Effect of CMT‐3 on total secreted protein levels in E‐10 CM. E‐10 Cells were cultured in 35S TransLabel‐containing SFM overnight. CM was collected and electrophoresed on SDS‐PAGE. The gel was dried and exposed to X‐ray film.
Effect of CMT‐3 on cell viability
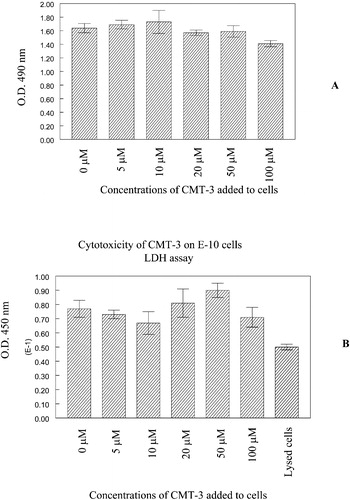
While the levels of the majority of the proteins released by E‐10 cells did not appear to be diminished after incubation in the presence of CMT‐3, we chose to evaluate the effect of this agent on viability of the cells at the end of a 48 hour incubation under conditions which might model a stromal environment, especially in the light of the previous report on the response of prostate tumour cell lines to CMT‐3 indicating that this agent induces cytotoxicity as well as markers of apoptotic cell death Citation14. illustrates the effects of doses of CMT‐3 up to 100 µM on viability of the E‐10 cell line. There was no diminution in cell density or significant loss of viability of the E‐10 cells after the same duration of exposure to doses as high as 50 µM, and only modest reduction in cell density or loss of viability at 100 µM as assessed by MTS () and LDH () assays. Clearly, the concentration of CMT‐3, 10 and 20 µM, used in the current study had no effect on cell viability ().
Figure 7 Cytotoxicity of CMT‐3 to E‐10 cells on R22 ECM. E‐10 cells were plated onto ECM‐coated wells in the presence of different concentrations of CMT‐3 as described in Methods section. At the end of a 48‐hour incubation, cell viability was analyzed by MTS (A) or LDH (B) assay. Further details are described in the Methods section.
Discussion
Cancer invasion can be viewed as a tissue destructive process that involves proteolytic degradation of ECM. A major step in the sequence of events that constitute tumour invasion and subsequent metastatic spread is the interaction of the tumour cell with the ECM Citation19, Citation20. After tumour cells attach to the ECM, a variety of proteinases, in particular basement membrane degrading MMP‐9, are expressed, activated, and released to the surrounding environment so that migration of the cells through the proteolytically digested ECM is facilitated. Since most of the MMPs are secreted enzymes, matrix degradation was detected when conditioned medium alone collected from E‐10 cells cultured on plastic was added to ECM. However, much higher ECM degradative activity was detected when viable E‐10 cells were present on the matrix. E‐10 is a subclone isolated from a transfection of the MDA‐MB 231 breast cancer cell line with multiple copies of the MMP‐9 gene Citation15. MDA‐MB 231 cells alone only degraded about 6% of the ECM. Therefore, our results suggested that MMP‐9 played an important role in ECM degradation mediated by E‐10 cells. This increase in ECM degradative activity is due, at least in part, to increased activation of MMP‐9 when the cells are in contact with the matrix. It has become widely recognized that enhancement of tumour cell invasiveness is only one of many examples of modification of the cellular phenotype which may result from interactions with ECM. Cell adhesion molecules play an important role in modulation of cell functions by the ECM through their capacity to act as transducers from the exterior to the interior of the cell. Interactions of cells with matrix proteins mediated through surface receptors, such as the family of integrins, have been demonstrated Citation21, Citation22. Enhancement of MMP activity as well as expression by cooperatively interacting integrin‐mediated cell‐ECM interactions has also been reported Citation23, Citation24. Such multiple cell‐matrix interactions may be critical for mediating the enhanced activation of MMP‐9 which we observed in the conditioned medium from E‐10 cells cultured on ECM but not from cells cultured on plastic.
MMPs appear to play an important role in E‐10‐mediated ECM degradation, as evidenced in part by the capacity of CMT‐3 (a potent tetracycline‐based MMP inhibitor Citation10, Citation17) to inhibit matrix solubilization by E‐10 cells and their conditioned media. The diminution in ECM‐degrading activity of E‐10 cells cultured in the presence of CMT‐3 may be ascribed to reduced extracellular levels as well as inhibited enzyme activity of MMP‐9 released by the breast carcinoma‐derived cell line, but, in contrast to the behaviour of prostate cell lines reported by Lokeshwar et al. Citation14, is not due to cytostasis or cytotoxicity.
The four rings of the tetracyclines, and especially the carbonyl and hydroxyl groups at C‐11 and C‐12, have been shown to constitute the minimum structural requirements for retention of MMP‐inhibitory activity of this family of drugs Citation10. CMTs probably inhibit gelatinolytic activity through binding to, or chelation of, Zn++ metal ions which are necessary for catalytic activity at the active site, as well as secondary Ca++ and Zn++ which are necessary for maintenance of the native three‐dimensional structure of the MMPs. Noteworthy, the therapeutically attainable Citation10 CMT‐3 concentrations (10 µM and 20 µM) significantly reduced but did not completely inhibit the gelatinase activity. However, the mechanism through which CMT‐3 reduces extracellular levels of MMP‐9 remains unclear. We have ruled out the possibility that CMT‐3 causes a general inhibition of protein synthesis. The diminution of MMP‐9 levels in the presence of CMT‐3 is not a reflection of a more general effect of this agent on total protein levels. It has a selective effect on MMP‐9 levels, without altering TIMP‐1 levels, so that CMT‐3 reduces the MMP‐TIMP ratio and shifts the proteinase‐antiproteinase balance in a less invasive direction. Brandt's laboratory has suggested that in the presence of doxycycline, the pro‐forms of some MMPs may undergo an abnormal series of processing events which results in premature denaturation followed by degradation and inactivation of the gelatinase enzyme Citation25. However, in our findings, no significant immunoreactive species smaller in molecular weight than the latent and active forms of MMP‐9 were detected on Western blots of conditioned medium from E‐10 cells or their parental MDA‐MB‐231 line cultured in the presence of CMT‐3.
Previous reports have suggested that CMTs can inhibit MMP mRNA transcription Citation26. Modulation of expression of MMPs can be also achieved through modulation of intracellular regulatory pathways which control expression of cytokines and growth factors. Previous studies have shown that interleukin‐1 (IL‐1), tumour necrosis factor‐α (TNF‐α), interferon‐γ ( IFN‐γ), and transforming growth factor‐β (TGF‐β) may all regulate synthesis of MMPs in the local environment Citation3–7. Among all these cytokines, TNF‐α has been reported to trigger an increase in MMP‐9 levels Citation27. Our results indicate that levels of soluble TNF‐α, as well as MMP‐9, released by E‐10 cells were diminished in the presence of CMT‐3. Soluble TNF‐α is shed from its transmembrane protein precursor through proteolytic cleavage mediated by TNF‐α converting enzyme (TACE). Thus, it may be speculated that since CMT‐3 is a broad spectrum and pleiotropic MMP‐inhibitor Citation10, Citation28–33, it may inhibit TACE activity, although the effect observed could also be due to inhibition of TNF‐α synthesis. This diminution may contribute to the reduction in MMP‐9 levels as a consequence of a weakened autocrine activation signal. However, membrane‐bound TNF‐α is also capable of binding to TNF‐α receptors and initiating a signal transduction pathway through cell‐cell interactions, so the diminution in levels of the MMP and the inflammatory cytokine may be correlative rather than causal. However, this is a very preliminary speculation and needs to be further investigated. At this time, a number of possible steps in transcription, translation, or secretion ranging from diminished synthesis of mRNA or protein to increased destruction of either macromolecule are still possible targets for the pleiotropic action of CMT‐3. However, it seems that the effects on MMP‐9 expression we found here are not likely to involve a single specific transcriptional regulatory mechanism, since the promoter for the transfected MMP‐9 genes in the E‐10 cells is of viral origin, rather than the natural promoter. Moreover, the diminution in MMP‐9 released by non‐transfected MDA‐MB‐231 cells cultured in the presence of CMT‐3 indicates that the results with the E‐10 cells cannot be ascribed to an idiosyncratic effect of CMT‐3 on the transfected line. In recent investigations we have undertaken on other breast carcinoma‐derived cell lines, which are not transfected to overproduce any MMPs, we have observed similar diminutions in levels of MMP‐9 and vascular endothelial growth factor (VEGF) which were released into the supernatant medium of cells cultured in the presence of CMT‐3, again in the absence of cytotoxicity. As a result of its multiple activities, CMT‐3 may offer promise for suppression of the invasive/metastatic phenotype of tumours, and when used in conjunction with other chemotherapy agents, may be incorporated into more successful cancer treatment.
Moreover, recent studies have shown that minimal or normal, but not pathologically excessive, levels of MMPs exert physiological protective or defensive host functions such as processing of growth factors, chemokines, cytokines and endogenous proteinase inhibitors (i.e. serpins) Citation8, Citation9, Citation33. Therefore, instead of excessively potent MMP‐inhibitors Citation33 the less efficient MMP‐inhibitors, such as CMT‐3 Citation10, can be one of the future type of drugs that only reduces the pathologically‐excessive MMPs without abolishing the protective host responses Citation8, Citation10, Citation33. These multiple effects of CMT‐3 may offer promise for use in suppressing tumour invasion, and if used in conjunction with other chemotherapy agents, may lead to more successful treatment of cancer.
Acknowledgements
The research in our laboratories has been supported by grants from the National Institute of Dental and Craniofacial Research (R01‐DE‐10985), the New York State Office of Science and Technology (SUNY Stony Brook Center for Biotechnology), the Center of Excellence Program of the Academy of Finland and TEKES, the Helsinki University Research Funds, the HUCH‐EVO (TYH 3306, TYH 2267, TI 020 Y 0002 and TYH 5306) grants, the Wilhelm and Else Stockmann Foundation, the Academy of Finland (T. Sorsa), the Invalid Foundation, the Finska Läkaresällskapet and the CollaGenex Pharmaceuticals, Inc. Y. Gu is a Predoctoral Dental Scientist Fellow of the NIDCR (F30‐DE05741).
References
- Heppner K. J., Matrisian L. M., Jensen R. A., Rodgers W. H. Expression of most matrix 1 metalloproteinase family members in breast cancer represents a tumor‐induced host response. Am J Pathol 1996; 149: 273–82
- Dickson R. B., Shi Y. E., Johnson M. D. Matrix‐degrading proteases in hormone‐dependent breast cancer. Breast Cancer Res Treat 1994; 31: 167–73
- Shinmei M., Masuda K., Kikuchi T., Shimomura Y. J. The role of cytokines in chondrocyte mediated cartilage degradation. J Rheumatol Suppl 1989; 18: 32–4
- Duncan M. R., Berman B. Differential regulation of glycosaminoglycan, fibronectin, and collagenase production in cultured human dermal fibroblasts by interferon‐alpha, ‐beta, and ‐gamma. Arch Dermatol Res 1989; 281: 11–18
- Vollberg T. M., George M. D., Jetten A. M. Induction of extracellular matrix gene expression in normal human keratinocytes by transforming growth factor beta is altered by cellular differentiation. Exp Cell Res 1991; 193: 93–00
- Ahmadzadeh N., Shingu M., Nobunaga M. The effect of recombinant tumor necrosis factor‐alpha on superoxide and metalloproteinase production by synovial cells and chondrocytes. Clin Exp Rheumatol 1990; 8: 387–91
- Unemori E. N., Hibbs M. S., Amento E. P. Constitutive expression of a 92‐kD gelatinase (type V collagenase) by rheumatoid synovial fibroblasts and its induction in normal human fibroblasts by inflammatory cytokines. J Clin Invest 1991; 88: 1656–62
- Owen C. A., Hu Z., Lopez‐Otin C., Shapiro S. D. Membrane‐bound matrix metalloproteinase‐8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase‐resistant collagenases and serpinase. J Immunol 2004; 172: 7791–803
- Sorsa T., Golub L. M. Is the excessive inhibition of matrix metalloproteinases (MMPs) by potent synthetic MMP inhibitors (MMPIs) desirable in periodontitis and other inflammatory diseases? ie., “leaky” MMPIs vs. excessively efficient drugs. Oral Dis. 2005; accepted for publication
- Golub L. M., Lee H. M., Ryan M. E., Giannobile W. V., Payne J., Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non‐antimicrobial mechanisms. Adv Dent Res 1998; 12: 12–26
- Zucker S., Lysik R. M., Ramamurthy N. S., Golub L. M., Wieman J. M., Wilkie D. P. Diversity of melanoma plasma membrane proteinases: inhibition of collagenolytic and cytolytic activities by minocycline. J Natl Cancer Inst 1985; 75: 517–25
- Zucker S., Wieman J., Lysik R. M., Imhof B., Nagase H., Ramamurthy N. S. Gelatin‐degrading type IV collagenase isolated from human small cell lung cancer. Invasion Metastasis 1989; 9: 167–81
- Rifkin B. R., Vernillo A. T., Golub L. M., Ramamurthy N. S. Modulation of bone resorption by tetracyclines. Ann N Y Acad Sci 1994; 732: 165–80
- Lokeshwar B. L., Selzer M. G., Zhu B. Q., Block N. L., Golub L. M. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non‐antimicrobial tetracycline analog (COL‐3) in a metastatic prostate cancer model. Int J Cancer 2002; 98: 297–309
- Ramos‐DeSimone N., Hahn‐Dantona E., Sipley J., Nagase H., French D. L., Quigley J. P. Activation of matrix metalloproteinase‐9 (MMP‐9) via a converging plasmin/stromelysin‐1 cascade enhances tumor cell invasion. J Biol Chem 1999; 274: 13066–76
- Rinehart A. R., Mallya S., Simon S. R. Human alpha 1‐proteinase inhibitor binds to extracellular matrix in vitro. Am J Respir Cell Mol Biol 1993; 9: 666–79
- Gu Y., Lee H. M., Roemer E. J., Musacchia L., Golub L. M., Simon S. R. Inhibition of tumor cell invasiveness by chemically modified tetracyclines. Curr Med Chem 2001; 8: 261–70
- Roemer E. J., Stanton K., Simon S. R. In vitro assay systems for cell interactions with intersititial extracellular matrix. In Vitro Toxicology 1994; 7: 209–24
- Fidler I. J., Radinsky R. J. Genetic control of cancer metastasis. J Natl Cancer Inst 1990; 82: 166–8
- Liotta L. A., Steeg P. S., Stetler‐Stevenson W. G. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 1991; 64: 327–36
- Akiyama S. K., Nagata K., Yamada K. M. Cell surface receptors for extracellular matrix components. Biochim Biophys Acta 1990; 1031: 91–110
- Hynes R. O. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992; 69: 11–25
- Kozlova N. I., Morozevich G. E., Soloveyva N. I., Vinokurova S. V., Berman A. E. Integrin expression and collagenase activity of RSV‐transformed Syrian hamster fibroblasts, differing in spontaneous metastasizing. Biochem Mol Biol Int 1997; 43: 529–39
- Langholz O., Rockel D., Mauch C., Kozlowska E., Bank I., Krieg T. Collagen and collagenase gene expression in three‐dimensional collagen lattices are differentially regulated by alpha 1 beta 1 and alpha 2 beta 1 integrins. Cell Biol 1995; 131: 1903–15
- Smith G. N., Brandt K. D., Hasty K. A. Procollagenase is reduced to inactive fragments upon activation in the presence of doxycycline. Ann N Y Acad Sci 1994; 732: 436–8
- Uitto V. J., Firth J. D., Nip L., Golub L. M. Doxycycline and chemically modified tetracyclines inhibit gelatinase A (MMP‐2) gene expression in human skin keratinocytes. Ann N Y Acad Sci 1994; 732: 140–51
- Mackay A. R., Ballin M., Pelina M. D., Farina A. R., Nason A. M., Hartzler J. L. Effect of phorbol ester and cytokines on matrix metalloproteinase and tissue inhibitor of metalloproteinase expression in tumor and normal cell lines. Invasion Metastasis 1992; 12: 168–84
- Gearing A. J., Beckett P., Christodoulou M., Churchill M., Clements J., Davidson. Processing of tumour necrosis factor‐alpha precursor by metalloproteinases. Nature 1994; 370: 555–7
- Mohler K. M., Sleath P. R., Fitzner J. N., Cerretti D. P., Alderson M., Kerwar S. S. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 1994; 370: 218–20
- Moss M. L., Jin S. L., Milla M. E., Bickett D. M., Burkhart W., Carter. Cloning of a disintegrin metalloproteinase that processes precursor tumour‐necrosis factor‐alpha. Nature 1997; 385: 733–6
- Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F. A metalloproteinase disintegrin that releases tumour‐necrosis factor‐alpha from cells. Nature 1997; 385: 729–33
- Amour A., Slocombe P. M., Webster A., Butler M., Knight C. G., Smith B. J. TNF‐alpha converting enzyme (TACE) is inhibited by TIMP‐3. FEBS Lett 1998; 435: 39–44
- Overall C. M., Lopez‐Otin C. Strategies for MMP inhibition in cancer: innovations for the post‐trial. Nat Rev Cancer 2002; 2: 657–72