Abstract
Despite the fact that controversy remains around the underlying pathophysiological processes leading to the development of the metabolic syndrome (insulin resistance and/or hyperinsulinemia versus abdominal obesity), there is increased recognition that abdominal obesity is the most prevalent form of the metabolic syndrome. Although it has been well established that there is a greater prevalence of chronic metabolic diseases such as diabetes and cardiovascular diseases in obese patients than among normal weight individuals, obesity is a remarkably heterogeneous condition and not every obese patient is characterized by co‐morbidities. In this regard, body fat distribution, especially visceral adipose tissue accumulation, has been found to be a major correlate of a cluster of diabetogenic, atherogenic, prothrombotic and proinflammatory metabolic abnormalities referred to as the metabolic syndrome. Due to its anatomic location and peculiar metabolic, hyperlipolytic activity, the expanded visceral adipose depot is a key correlate of the altered cardiometabolic risk profile observed among individuals with a high‐risk abdominal obesity phenotype. Evidence suggests that this dysmetabolic profile is predictive of a substantially increased risk of coronary heart disease even in the absence of classical risk factors. Finally, a moderate weight loss in initially abdominally obese patients is associated with a preferential mobilization of visceral adipose tissue, which in turn leads to substantial improvements in the metabolic risk profile predictive of a reduced risk of coronary heart disease and of type 2 diabetes.
Introduction
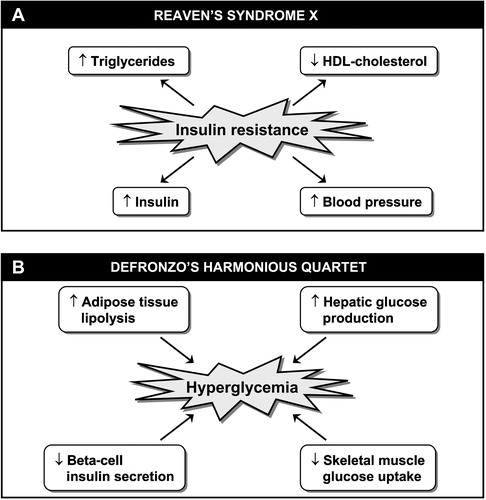
In 1988, Reaven Citation1 coined the term syndrome X and proposed that an impaired in vivo insulin action was a core component responsible for a cluster of abnormalities/risk factors which included compensatory hyperinsulinemia, hypertriglyceridemia, low high‐density lipoprotein (HDL)‐cholesterol levels and hypertension (). As the simultaneous presence of these metabolic abnormalities has been shown to increase the risk of coronary heart disease (CHD) even in the absence of diabetes or hyperglycemia Citation2, the introduction of such a concept was a remarkable advance in preventive medicine as it did emphasize the need to go beyond the assessment of conventional risk factors in order to identify high‐risk individuals. At the same time that Reaven's syndrome X was introduced, DeFronzo Citation3 underlined the importance of a triad of skeletal muscle, liver and beta‐cell abnormalities in the etiology of type 2 diabetes. He did emphasize that skeletal muscle insulin resistance was an essential element in the pathophysiology of type 2 diabetes. Under his model, an insulin resistant liver leading to an increased hepatic glucose production would contribute to explain the glucose intolerance of insulin resistant individuals. As insulin resistance can be frequently observed in non‐diabetic individuals, DeFronzo also emphasized that a relative deficit in beta‐cell insulin secretion capacity was essential to observe the conversion from an insulin resistant state to type 2 diabetes. Since it has now become obvious that an insulin resistant state is also accompanied by an impaired antilipolytic effect of insulin in adipose tissue leading to an overproduction of free fatty acids (FFA) Citation4,5, DeFronzo's ‘triumvirate’ concept has evolved to the notion of a ‘harmonious quartet’ Citation6 where skeletal muscle insulin resistance, increased hepatic glucose production, increased adipose tissue lipolysis and reduced beta‐cell insulin secretion capacity are all involved in the regulation of the hyperglycemic state of type 2 diabetes ().
Figure 1. A. Insulin resistance syndrome or syndrome X as initially described by Reaven in 1988. B. DeFronzo's ‘harmonious quartet’ showing abnormalities involved in the pathophysiology of type 2 diabetes.
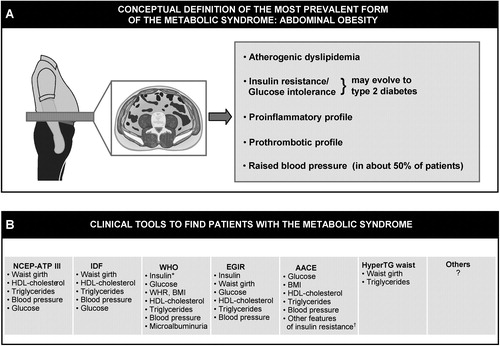
As insulin resistance can be measured in specialized clinical research settings even in the absence of hyperglycemia, and as there is evidence that markers of an insulin resistant state are predictive of an increased risk of CHD Citation7, there has been considerable interest in the study of the clustering abnormalities observed among insulin resistant individuals. Furthermore, the pathophysiology of insulin resistance has been the topic of excellent review papers Citation8,9 and the study of the possible molecular mechanisms involved in the etiology of insulin resistance has been and remains the subject of numerous investigations. Addressing this specific topic is beyond the scope of the present paper and the reader is referred to this previously published literature. As there is increased recognition that abdominal obesity is by far the most prevalent form of insulin resistance Citation10, the present paper will focus on the possible mechanisms linking abdominal obesity to insulin resistance and to its related cluster of atherogenic metabolic abnormalities which are now often referred to as the metabolic syndrome ().
Figure 2. Distinction to be made between the conceptual definition of the most prevalent form of the metabolic syndrome(abdominal obesity) (panel A) versus clinical tools that have been proposed by various organizations/groups to identify patients likely to have the metabolic syndrome (panel B). Although finding patients with the metabolic syndrome is important in clinical practice, how to integrate this information in global cardiovascular risk assessment remains an unresolved issue (see text). NCEP‐ATP III = National Cholesterol Education Program‐Adult Treatment Panel III; IDF = International Diabetes Federation; WHO = World Health Organization; EGIR = European Group for the study of Insulin Resistance; AACE = American Association of Clinical Endocrinologists; HyperTG waist = hypertriglyceridemic waist; BMI = body mass index; WHR = waist‐to‐hip ratio. *Insulin resistance measured under hyperinsulinemic euglycemic conditions. †Includes family history of type 2 diabetes, polycystic ovary syndrome, sedentary lifestyle, advancing age and ethnic groups susceptible to type 2 diabetes.
Key messages
Abdominal obesity is the most prevalent form of the metabolic syndrome.
Adipose tissue is also an endocrine organ which secretes adipokines involved in the atherogenic/diabetogenic metabolic risk profile of abdominal obesity.
Simple and discriminant tools must be developed to optimally assess the global cardiovascular risk associated with the presence of classical risk factors and markers of the metabolic syndrome (cardiometabolic risk).
Obesity, insulin resistance and the metabolic syndrome: Beyond body weight
Despite the well documented health hazards of obesity Citation11,12, physicians are constantly challenged by the absence of complications in some very obese patients who may nevertheless have a fairly normal metabolic risk factor profile despite their very significant obesity. On the other hand, some moderately overweight individuals are characterized by a whole cluster of atherogenic and diabetogenic metabolic abnormalities. In this regard, a whole series of imaging and metabolic studies conducted over almost two decades have revealed that, in both men and women, the amount of visceral adipose tissue, which can be reliably assessed by imaging techniques such as computed tomography, is a critical correlate of the presence of a substantially altered metabolic risk factor profile predictive of an ncreased risk for the development of type 2 diabetes and cardiovascular disease (CVD) Citation13–18. Therefore, although there is evidence that obesity increases the risk of chronic complications, there is also compelling evidence that viscerally obese patients represent the subgroup of obese subjects characterized by the most severe metabolic abnormalities.
Visceral obesity, insulin resistance and additional features of the metabolic syndrome: The portal theory and beyond
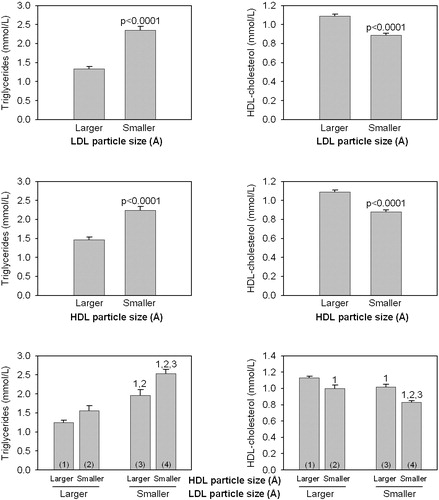
There is ample evidence indicating that an increased adipose tissue mass is associated with an impaired in vivo insulin action. Such association had led some investigators to suggest that subcutaneous and not visceral adiposity was the key correlate of insulin resistance Citation19–22. Although there is solid evidence that the small visceral adipose depot does not make a major contribution to systemic FFA levels Citation23, studies that have carefully matched subjects for total body fat (or for subcutaneous fat) but with high versus low levels of visceral adipose tissue have clearly shown that viscerally obese patients have the most disturbed metabolic risk profile Citation14,Citation24–26. Furthermore, the notion that subcutaneous fat is the culprit cannot be reconciled with the observation in clinical practice of very obese patients who have a normal metabolic risk profile when they have low levels of visceral adipose tissue Citation27–29. Thus, studies that have used imaging techniques to measure subcutaneous versus visceral adipose tissue accumulation have clearly shown that viscerally obese subjects (even in the absence of clinical obesity) represented the subgroup of individuals characterized by the most severe insulin resistant state Citation14,Citation24,25,Citation30. Because of the peculiar metabolism and anatomic location of the intra‐abdominal adipose tissue which is partly drained by the portal vein, an increased accumulation of hyperlipolytic visceral adipose tissue has therefore been associated with an impaired FFA metabolism (as these hypertrophied visceral adipocytes have been shown to be resistant to the antilipolytic effect of insulin and also characterized by an impaired FFA esterification) Citation31,32 (). Such flux of FFA toward the liver favors an increased secretion of triglyceride (TG)‐rich lipoproteins, reduced hepatic degradation of apolipoprotein B and of insulin leading to hyperapolipoprotein B and hyperinsulinemia, and to an increased hepatic glucose production contributing to glucose intolerance Citation33,34. In addition, it has been reported that viscerally obese patients are characterized by an exaggerated postprandial TG response Citation17 as the increased very low‐density lipoprotein (VLDL) levels associated with a fasting hypertriglyceridemic state compete with dietary chylomicrons for their lipolysis by the enzyme lipoprotein lipase which is saturated in the postprandial state. Such an elevated concentration of TG‐rich lipoproteins found in viscerally obese patients favors the transfer of TG from TG‐rich lipoproteins to low‐density lipoprotein (LDL) and HDL in exchange for cholesteryl esters located in LDL and HDL. This process leads to the TG‐enrichment of LDL and HDL particles which are then subjected to lipolysis by the enzyme hepatic lipase. There is some evidence suggesting that hepatic lipase activity is elevated in viscerally obese patients Citation35,36 and an increased activity of this enzyme promotes the formation of small LDL and HDL particles. As the core of LDL and HDL particles are relatively depleted in esterified cholesterol, the lipolysis of the core‐TG molecules thus leads to the formation of cholesteryl ester‐depleted, small LDL and HDL particles. Such process explains the increased proportion of small LDL and HDL particles in visceral obesity and is also largely responsible for the reduced HDL‐cholesterol concentration found in this condition Citation18,Citation37.Thus, we have reported that the small, dense LDL and HDL phenotype is also a component of the high TG, reduced HDL‐cholesterol dyslipidemia which is found in abdominally obese insulin resistant patients () Citation38. In this regard, plasma LDL‐cholesterol concentration is not increased in viscerally obese patients Citation15,Citation26. In addition, as the plasma LDL‐cholesterol concentration shows no correlation with LDL and HDL particle sizes Citation13,Citation38, clinicians should not rely on LDL‐cholesterol levels to identify patients likely to have small LDL or HDL particles. Thus, there is a metabolic rationale linking the expanded abdominal visceral depot to liver insulin resistance, hyperinsulinemia, glucose intolerance and to the high TG, low HDL‐cholesterol, small LDL dyslipidemia often observed in abdominally obese patients.
Figure 3. Working mechanistic model for the relation of excess visceral adipose tissue accumulation to the insulin resistant, dyslipidemic, prothrombotic and proinflammatory profile of abdominally obese patients.
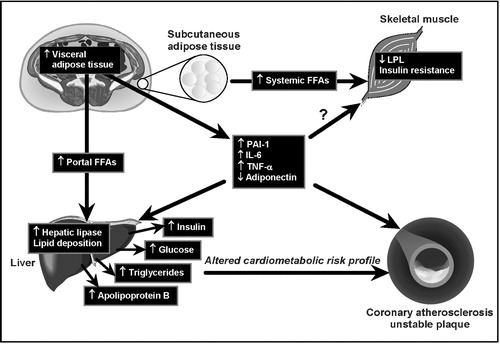
Figure 4. Triglyceride and HDL‐cholesterol levels in men according to LDL particle size (smaller<251.6 Å; larger ⩾251.6 Å) and HDL particle size (smaller<83.5 Å; larger ⩾83.5 Å). 1,2,3: significantly different from the corresponding subgroup P<0.0001. Thus, subjects with both small LDL and small HDL particles had the highest plasma triglycerides and the lowest HDL‐cholesterol concentrations.
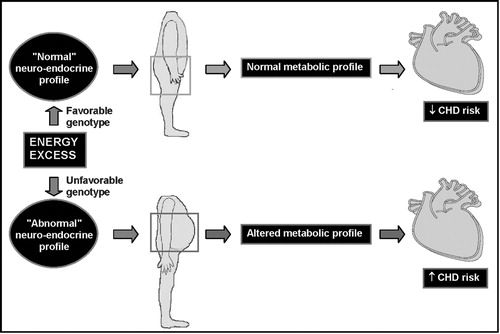
However, this ‘portal theory’ has often been questioned Citation39,40 and evidence has also been provided that such a model cannot reconcile all metabolic abnormalities found in abdominally obese patients. For example, even among viscerally obese patients, the expanded visceral adipose depot does not represent a large proportion of the total adipose tissue mass Citation39. Thus, even if visceral adipocytes have a hyperlipolytic state, a large percentage of adipose tissue‐derived fatty acids must be derived from the subcutaneous fat depots Citation41. Another possibility is that visceral obesity is a marker of the relative inability of subcutaneous fat to clear and accumulate the energy surplus originating from the diet Citation42. It has also been proposed that visceral obesity could be a marker of a more primary neuroendocrine abnormality preferentially partitioning the energy surplus in visceral rather than in subcutaneous adipose tissue Citation43,44. For instance, the higher glucocorticoid receptor density in visceral than subcutaneous fat cells could make the visceral depot exquisitely sensitive to an activated hypothalamic‐pituitary‐adrenal axis, leading to visceral adipose tissue accumulation Citation44,45. An obvious clinical example of this situation is Cushing's syndrome, in which patients have both high cortisol levels and expanded waistlines as well as evidence of marked insulin resistance Citation46. Steroid hormones are also obviously involved as estrogens increase peripheral fat accumulation in women whereas increased androgen levels appear to protect against visceral fat deposition in men Citation47. Metabolic and body fat distribution studies conducted on transsexuals who received hormone replacement therapy have clearly shown the remarkable impact of sex steroids on body fat distribution and related metabolic parameters Citation48,49. Thus, visceral obesity may be a marker of an abnormal neuro‐endocrine profile leading to both features of insulin resistance and to visceral obesity. However, once the visceral depot is expanded, the peculiar metabolic profile of the hypertrophied visceral adipocytes may exacerbate the patient's condition and the individual may enter a vicious cycle exacerbating his/her risk of diabetes and CHD ().
Figure 5. Hypothetical model by which visceral obesity is partly the consequence of a given neuro‐hormonal milieu preferentially partitioning the excess energy in the visceral fat depot rather than in subcutaneous adipose tissue.
Adipose tissue as an endocrine organ: The missing link?
It is also now recognized that adipose tissue is a remarkable endocrine organ which produces pro‐thrombotic and inflammatory molecules Citation50. Such ‘adipokines’ could contribute to exacerbate the patient's risk of diabetes and CHD. For instance, viscerally obese patients have been shown to have an impaired fibrinolysis and an increased susceptibility to thrombosis as well as evidence for the presence of a chronic inflammatory state Citation16, Citation51–56. Markedly elevated plasma C‐reactive protein (CRP) concentrations have been reported in abdominally obese patients, the highest levels being found in obese patients with a selective excess of visceral adipose tissue Citation16. Such elevated CRP levels may be the consequence of altered cytokine levels (elevated interleukin (IL)‐6 and tumor necrosis factor (TNF)‐α originating from the expanded adipose tissue) Citation57,58 promoting a chronic inflammatory state which could also contribute to the etiology of insulin resistance and CVD. Recently, another adipose tissue‐specific cytokine, adiponectin, has been reported to be reduced in abdominally obese patients Citation59,60. Such a low adiponectin concentration has been suggested to contribute to the development of insulin resistance and CHD in abdominally obese patients with the features of the metabolic syndrome Citation59,Citation61,61. Cross‐sectional studies have reported lower adiponectin levels in type 2 diabetic patients as well as in patients with documented CHD Citation63,64. Furthermore, a recent prospective study has shown that a low adiponectin concentration could predict an increased CHD risk beyond the contribution of classical CHD risk factors Citation65. Several mechanisms could explain the apparently favorable effects of adiponectin regarding its potential protection against type 2 diabetes and CHD and they are summarized in .
Table I. In vitro metabolic effects of adiponectin which may potentially protect against the development of type 2 diabetes and coronary heart disease.
Therefore, viscerally obese patients, even in the absence of classical CVD risk factors such as hypertension, hyperglycemia, elevated LDL‐cholesterol concentrations, could be at increased risk for an acute coronary syndrome due to the presence of an atherogenic, prothrombotic and proinflammatory profile Citation38. However, although several features of this metabolic syndrome have been shown to predict an increased CHD risk, we presently do not know which features of the metabolic syndrome are critical to assess in order to improve our ability to predict CHD events once traditional risk factors are taken into account. This issue is the topic of considerable debate in the literature Citation66. Additional studies are clearly warranted and this will be a fertile area of investigation in future studies.
The cluster of metabolic abnormalities of visceral obesity: Impact on CHD risk
Over the years, attempts have been made to quantify the risk of CHD associated with the features of the metabolic syndrome that we and others have found in abdominally obese men, even in the absence of type 2 diabetes. Several prospective studies have suggested that fasting hyperinsulinemia, as a crude marker of insulin resistance in non‐diabetic individuals, was predictive of an increased CHD risk Citation67–71. In the sample of 2103 asymptomatic middle‐aged men of the Québec Cardiovascular Study followed for incidence of a first CHD event over 5 years, we found that some of the metabolic features of abdominal obesity, namely fasting hyperinsulinemia (as a crude marker of insulin resistance), elevated apolipoprotein B concentration, small LDL particles, an elevated cholesterol/HDL‐cholesterol ratio, and an elevated CRP concentration (a marker of low chronic inflammation) were all predictive of an increased risk of CHD Citation68,Citation72–75. We also found that the simultaneous presence of these features of the metabolic syndrome were predictive of a markedly elevated risk of CHD Citation7. As some of these markers are not well accepted CHD risk factors in current medical practice and in national guidelines, we have compared the ability of some of these metabolic markers of the insulin resistance syndrome (the atherogenic metabolic triad of elevated insulin and apolipoprotein B levels and small LDL particles) to predict CHD risk as opposed to commonly used algorithms such as the Framingham risk chart Citation76. Not surprisingly, we found that a high Framingham score was predictive of an increased risk of CHD in men of the Québec Cardiovascular Study Citation76. However, even in the absence of classical risk factors (and therefore among men with a low Framingham score), we found the features of the metabolic syndrome to be predictive also of an equally increased risk of CHD Citation76. Therefore, these results indicate that our current risk assessment algorithm based on Framingham risk factors does not always identify high‐risk individuals with the features of the metabolic syndrome if these individuals do not have traditional risk factors such as hypertension, smoking, elevated cholesterol or type 2 diabetes. There is therefore a need to further refine current CHD risk assessment algorithms by incorporating critical markers of the metabolic syndrome. However, which combination of markers would provide the optimal discrimination of high‐risk individuals at the lowest cost while being easily standardized remains an unresolved issue.
This is essentially the rationale which was used to develop the ‘hypertriglyceridemic waist’ concept Citation77 as there was a need to implement in clinical practice a simple screening approach which could help primary care physicians to identify, in a cost‐effective manner, the subgroup of abdominally obese patients with the atherogenic features of the metabolic syndrome. ‘Hypertriglyceridemic waist’ must also be discussed in the context of guidelines published to identify patients likely to have the metabolic syndrome. For instance, the National Cholesterol Education Program‐Adult Treatment Panel III (NCEP‐ATP III) guidelines Citation78 have also acknowledged the importance of considering waist girth and fasting TG as two key elements to identify individuals with the metabolic syndrome although different cutoff values were proposed. Additional work will be required to validate the screening tools proposed by various organizations. It is recommended that inclusion of variables and selection of cutoff values be based on hard clinical events. Lack of validation of clinical criteria of the metabolic syndrome against ‘hard’ CHD events is one of the criticisms addressed Citation66 to the metabolic syndrome criteria/cutoffs proposed by current guidelines Citation77–82 (). Furthermore, once global CHD risk is assessed by taking into account ‘classical’ risk factors, it has been claimed that whether the presence/absence of clinical criteria of the metabolic syndrome would alter clinical management of these patients is not clear Citation83. However, once the notion that the most prevalent form of the metabolic syndrome is abdominal obesity, such a concept emphasizes the need to assess and target abdominal obesity, an objective poorly achieved in current clinical practice.
Targeting weight loss in high‐risk abdominally obese patients with the metabolic syndrome: Recent evidence and unanswered questions
Despite the evidence that abdominal obesity characterized by an excess of visceral adipose tissue and related features of the metabolic syndrome is a high‐risk obesity phenotype, no study has shown that aiming at visceral adipose tissue loss as a therapeutic target would reduce the risk of CHD and of type 2 diabetes in patients characterized by this condition. The best data available on the long‐term benefits of moderate weight loss come from two landmark trials that were recently published: the Finnish Prevention of Diabetes study Citation84 and the American Diabetes Prevention Program Citation85. Both trials recruited non‐diabetic patients with impaired glucose tolerance (IGT) who were randomized either to a lifestyle modification program aiming at moderate weight loss (produced by a combination of increased physical activity and moderate reduction of caloric intake) or to a placebo group receiving usual medical care. In the American Diabetes Prevention Program, there was also another group who received metformin. Both trials were remarkably similar in showing that moderate weight loss induced by the lifestyle modification program reduced the incidence of diabetes by 58%. Furthermore, the American Diabetes Prevention Program showed that the risk of conversion from IGT to diabetes was reduced to a greater extent (58%) by lifestyle than by metformin (31%). Another trial that used a weight loss agent (orlistat) also reported the added benefits of pharmacotherapy of obesity beyond a lifestyle modification program in reducing the risk of developing type 2 diabetes over 4 years, such benefits being particularly important among obese patients with IGT baseline Citation86.
The above three trials provide evidence that moderate weight loss produced by an improved diet and a more physically active lifestyle (without/with pharmacotherapy) can indeed decrease the risk of developing type 2 diabetes in a high‐risk population with IGT. However, although the contribution of these studies is remarkable in showing that prevention can work and in providing evidence on the type of support and of facilities needed to successfully induce moderate weight loss in subjects at high risk for type 2 diabetes, several questions remain unanswered. Although no computed tomography data on visceral adiposity are available, subjects enrolled in the Finnish Prevention of Diabetes study and the American Diabetes Prevention Program were abdominally obese. Thus, was the substantial reduction in risk of developing diabetes related to a selective loss of visceral adipose tissue? Was the assumed loss of visceral adipose tissue maintained throughout the two trials? Would the loss of visceral adipose tissue induced by moderate weight loss be maintained once a reduced body weight is achieved and maintained over time? Did moderate weight loss prevent or just retard conversion to diabetes? Is such a moderate weight loss sufficient to substantially improve the features of the metabolic syndrome in patients with IGT? Would the assumed improvements in metabolic risk factors produced by these lifestyle modification programs be sustained had the reduced body weight been maintained over time?
Clearly, a moderate weight loss of only 5%–10% is associated with greater metabolic benefits than what could be explained by such fairly limited weight loss which does not normalize body weight in most obese patients Citation87,88. There is a possible explanation for this phenomenon. For instance, we and others have shown that weight loss is associated with a preferential mobilization of visceral adipose tissue compared to subcutaneous fat, especially among patients with a high accumulation of visceral adipose tissue at baseline Citation88–93. From the close link between visceral obesity and metabolic complications, such a loss of visceral fat may generate substantial improvements in the metabolic risk factor profile, a concept concordant with results of Goodpaster et al. Citation90 and of Ross et al. Citation94,95 who have shown that the improvements in insulin sensitivity/glucose tolerance in response to weight loss were more closely related to the loss of visceral adipose tissue than to the reduction in total body fat or subcutaneous fat.
In summary, there is considerable evidence that the most prevalent form of the metabolic syndrome is associated with abdominal obesity, especially when accompanied by a selective deposition of visceral adipose tissue. This notion has important clinical implications as it does recognize the importance of identifying the expanded waist girth of metabolic syndrome patients as a therapeutic target for the optimal management of their CHD risk. Although most physicians have heard about the metabolic syndrome and now recognize its importance, they are nevertheless confused on how to detect patients with the metabolic syndrome, how to optimally assess global CHD risk and how to manage these patients, with proper identification of key variables and targets. The current debate about the relevance of considering the metabolic syndrome in clinical practice Citation66,Citation83 highlights the fact that physicians often confuse pathophysiology and clinical screening tools. While a considerable amount of additional research is needed to understand which are the key features of the metabolic syndrome that really add to global CHD risk as assessed by classical risk factors, simple and discriminant tools must be developed and provided to clinicians as there is an urgent need to implement efficient screening strategies to identify high‐risk abdominally obese patients.
Finally, despite our remarkable progress in our ability to treat CVD risk factors by pharmacotherapy, our clinical efficacy is quite limited and there is evidence that a substantial proportion of treated patients remain at high risk for an acute coronary event. It is proposed that until abdominal obesity is identified as a key therapeutic target, we will not optimally manage the risk of CVD in a clinically significant proportion of high‐risk patients Citation96.
Acknowledgments
Dr. Després' work has been supported by research grants from the Canadian Institutes of Health Research and the Canadian Diabetes Association.
References
- Reaven G. M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595–607
- Alexander C. M., Landsman P. B., Teutsch S. M., Haffner S. M. NCEP‐defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 2003; 52: 1210–4
- DeFronzo R. A. Lilly lecture 1987. The triumvirate: beta‐cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667–87
- Björntorp P. Abdominal obesity and the development of non‐insulin dependent diabetes mellitus. Diabetes Metab Rev 1988; 4: 615–22
- Kissebah A. H., Peiris A. N. Biology of regional body fat distribution: Relationship to non‐insulin‐dependent diabetes mellitus. Diabetes Metab Rev 1989; 5: 83–109
- Bajaj M., Defronzo R. A. Metabolic and molecular basis of insulin resistance. J Nucl Cardiol 2003; 10: 311–23
- Lamarche B., Tchernof A., Mauriège P., Cantin B., Dagenais G. R., Lupien P. J., et al. Fasting insulin and apolipoprotein B levels and low‐density lipoprotein particle size as risk factors for ischemic heart disease. JAMA 1998; 279: 1955–61
- DeFronzo R. A., Ferrannini E. Insulin resistance: A multifaced syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991; 14: 173–94
- Reaven G. M. Pathophysiology of insulin resistance in human disease. Physiol Rev 1995; 75: 473–86
- Grundy S. M., Brewer H. B., Jr, Cleeman J. I., Smith S. C., Jr, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004; 109: 433–8
- Kissebah A. H., Freedman D. S., Peiris A. N. Health risk of obesity. Med Clin North Am 1989; 73: 111–38
- Bray G. A. Complications of obesity. Ann Intern Med 1985; 103: 1052–62
- Pascot A., Lemieux I., Prud'homme D., Tremblay A., Nadeau A., Couillard C., et al. Reduced HDL particle size as an additional feature of the atherogenic dyslipidemia of abdominal obesity. J Lipid Res 2001; 42: 2007–14
- Pouliot M. C., Després J. P., Nadeau A., Moorjani S., Prud'homme D., Lupien P. J., et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes 1992; 41: 826–34
- Tchernof A., Lamarche B., Prud'homme D., Nadeau A., Moorjani S., Labrie F., et al. The dense LDL phenotype. Association with plasma lipoprotein levels, visceral obesity, and hyperinsulinemia in men. Diabetes Care 1996; 19: 629–37
- Lemieux I., Pascot A., Prud'homme D., Alméras N., Bogaty P., Nadeau A., et al. Elevated C‐reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol 2001; 21: 961–7
- Couillard C., Bergeron N., Prud'homme D., Bergeron J., Tremblay A., Bouchard C., et al. Postprandial triglyceride response in visceral obesity in men. Diabetes 1998; 47: 953–60
- Després J. P., Moorjani S., Lupien P. J., Tremblay A., Nadeau A., Bouchard C. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis 1990; 10: 497–511
- Garg A. Regional adiposity and insulin resistance. J Clin Endocrinol Metab 2004; 89: 4206–10
- Abate N., Garg A., Peshock R. M., Stray Gundersen J., Adams Huet B., Grundy S. M. Relationship of generalized and regional adiposity to insulin sensitivity in men with NIDDM. Diabetes 1996; 45: 1684–93
- Goodpaster B. H., Thaete F. L., Simoneau J. A., Kelley D. E. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes 1997; 46: 1579–85
- Tai E. S., Lau T. N., Ho S. C., Fok A. C., Tan C. E. Body fat distribution and cardiovascular risk in normal weight women. Associations with insulin resistance, lipids and plasma leptin. Int J Obes Relat Metab Disord 2000; 24: 751–7
- Nielsen S., Guo Z., Johnson C. M., Hensrud D. D., Jensen M. D. Splanchnic lipolysis in human obesity. J Clin Invest 2004; 113: 1582–8
- Ross R., Freeman J., Hudson R., Janssen I. Abdominal obesity, muscle composition, and insulin resistance in premenopausal women. J Clin Endocrinol Metab 2002; 87: 5044–51
- Bacha F., Saad R., Gungor N., Janosky J., Arslanian S. A. Obesity, regional fat distribution, and syndrome X in obese black versus white adolescents: race differential in diabetogenic and atherogenic risk factors. J Clin Endocrinol Metab 2003; 88: 2534–40
- Després J. P., Moorjani S., Ferland M., Tremblay A., Lupien P. J., Nadeau A., et al. Adipose tissue distribution and plasma lipoprotein levels in obese women. Importance of intra‐abdominal fat. Arteriosclerosis 1989; 9: 203–10
- Wolf A. M., Beisiegel U., Kortner B., Kuhlmann H. W. Does gastric restriction surgery reduce the risks of metabolic diseases?. Obes Surg 1998; 8: 9–13
- Dixon J. B., O'Brien P. A disparity between conventional lipid and insulin resistance markers at body mass index levels greater than 34 kg/m(2). Int J Obes Relat Metab Disord 2001; 25: 793–7
- Adami G. F., Ravera G., Marinari G. M., Camerini G., Scopinaro N. Metabolic syndrome in severely obese patients. Obes Surg 2001; 11: 543–5
- Després J. P., Nadeau A., Tremblay A., Ferland M., Moorjani S., Lupien P. J., et al. Role of deep abdominal fat in the association between regional adipose tissue distribution and glucose tolerance in obese women. Diabetes 1989; 38: 304–9
- Marette A., Mauriège P., Marcotte B., Atgié C., Bouchard C., Thériault G., et al. Regional variation in adipose tissue insulin action and GLUT4 glucose transporter expression in severely obese premenopausal women. Diabetologia 1997; 40: 590–8
- Zierath J. R., Livingston J. N., Thorne A., Bolinder J., Reynisdottir S., Lonnqvist F., et al. Regional difference in insulin inhibition of non‐esterified fatty acid release from human adipocytes: relation to insulin receptor phosphorylation and intracellular signalling through the insulin receptor substrate‐1 pathway. Diabetologia 1998; 41: 1343–54
- Adeli K., Taghibiglou C., Van Iderstine S. C., Lewis G. F. Mechanisms of hepatic very low‐density lipoprotein overproduction in insulin resistance. Trends Cardiovasc Med 2001; 11: 170–6
- Arner P. Free fatty acids–do they play a central role in type 2 diabetes?. Diabetes Obes Metab 2001; 3(Suppl 1)S11–9
- Després J. P., Ferland M., Moorjani S., Nadeau A., Tremblay A., Lupien P. J. Role of hepatic‐triglyceride lipase activity in the association between intra‐abdominal fat and plasma HDL cholesterol in obese women. Arteriosclerosis 1989; 9: 485–92
- Carr M. C., Hokanson J. E., Zambon A., Deeb S. S., Barrett P. H., Purnell J. Q., et al. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J Clin Endocrinol Metab 2001; 86: 2831–7
- Onat A., Avci G. S., Barlan M. M., Uyarel H., Uzunlar B., Sansoy V. Measures of abdominal obesity assessed for visceral adiposity and relation to coronary risk. Int J Obes Relat Metab Disord 2004; 28: 1018–25
- Després J. P. Health consequences of visceral obesity. Ann Med 2001; 33((8))534–41
- Miles J. M., Jensen M. D. Counterpoint: visceral adiposity is not causally related to insulin resistance. Diabetes Care 2005; 28: 2326–8
- Frayn K. N. Visceral fat and insulin resistance–causative or correlative?. Br J Nutr 2000; 83(Suppl 1)S71–7
- Jensen M. D. Lipolysis: contribution from regional fat. Annu Rev Nutr 1997; 17: 127–39
- Lemieux I. Energy partitioning in gluteal‐femoral fat: does the metabolic fate of triglycerides affect coronary heart disease risk?. Arterioscler Thromb Vasc Biol 2004; 24: 795–7
- Björntorp P. Metabolic implications of body fat distribution. Diabetes Care 1991; 14((12))1132–43
- Björntorp P. Do stress reactions cause abdominal obesity and comorbidities?. Obes Rev 2001; 2: 73–86
- Rebuffe‐Scrive M., Walsh U. A., McEwen B., Rodin J. Effect of chronic stress and exogenous glucocorticoids on regional fat distribution and metabolism. Physiol Behav 1992; 52: 583–90
- Colao A., Pivonello R., Spiezia S., Faggiano A., Ferone D., Filippella M., et al. Persistence of increased cardiovascular risk in patients with Cushing's disease after five years of successful cure. J Clin Endocrinol Metab 1999; 84: 2664–72
- Tchernof A., Poehlman E. T. Effects of the menopause transition on body fatness and body fat distribution. Obes Res 1998; 6: 246–54
- Elbers J. M., Asscheman H., Seidell J. C., Gooren L. J. Effects of sex steroid hormones on regional fat depots as assessed by magnetic resonance imaging in transsexuals. Am J Physiol 1999; 276((2 Pt 1))E317–25
- Elbers J. M., Giltay E. J., Teerlink T., Scheffer P. G., Asscheman H., Seidell J. C., et al. Effects of sex steroids on components of the insulin resistance syndrome in transsexual subjects. Clin Endocrinol (Oxf) 2003; 58: 562–71
- Flier J. S. The adipocyte: storage depot or node on the energy information superhighway?. Cell 1995; 80: 15–8
- Juhan‐Vague I., Morange P., Renucci J. F., Alessi M. C. Fibrinogen, obesity and insulin resistance. Blood Coagul Fibrinolysis 1999; 10((Suppl 1))S25–8
- Juhan‐Vague I., Alessi M. C. Plasminogen activator inhibitor 1 and atherothrombosis. Thromb Haemost 1993; 70: 138–43
- Juhan‐Vague I., Pyke S. D., Alessi M. C., Jespersen J., Haverkate F., Thompson S. G. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European Concerted Action on Thrombosis and Disabilities. Circulation 1996; 94: 2057–63
- Juhan‐Vague I., Alessi M. C., Vague P. Thrombogenic and fibrinolytic factors and cardiovascular risk in non‐ insulin‐dependent diabetes mellitus. Ann Med 1996; 28: 371–80
- Juhan‐Vague I., Alessi M. C. PAI‐1, obesity, insulin resistance and risk of cardiovascular events. Thromb Haemost 1997; 78: 656–60
- Juhan‐Vague I., Alessi M. C. Regulation of fibrinolysis in the development of atherothrombosis: role of adipose tissue. Thromb Haemost 1999; 82: 832–6
- Yudkin J. S., Stehouwer C. D., Emeis J. J., Coppack S. W. C‐reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue?. Arterioscler Thromb Vasc Biol 1999; 19: 972–8
- Hak A. E., Stehouwer C. D., Bots M. L., Polderman K. H., Schalkwijk C. G., Westendorp I. C., et al. Associations of C‐reactive protein with measures of obesity, insulin resistance, and subclinical atherosclerosis in healthy, middle‐aged women. Arterioscler Thromb Vasc Biol 1999; 19: 1986–91
- Côté M., Mauriège P., Bergeron J., Alméras N., Tremblay A., Lemieux I., et al. Adiponectinemia in visceral obesity: impact on glucose tolerance and plasma lipoprotein and lipid levels in men. J Clin Endocrinol Metab 2005; 90: 1434–9
- Motoshima H., Wu X., Mahadev K., Goldstein B. J. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem Biophys Res Commun 2004; 315: 264–71
- Ukkola O., Santaniemi M. Adiponectin: a link between excess adiposity and associated comorbidities?. J Mol Med 2002; 80: 696–702
- Yamamoto Y., Hirose H., Saito I., Tomita M., Taniyama M., Matsubara K., et al. Correlation of the adipocyte‐derived protein adiponectin with insulin resistance index and serum high‐density lipoprotein‐cholesterol, independent of body mass index, in the Japanese population. Clin Sci (Lond) 2002; 103: 137–42
- Hotta K., Funahashi T., Arita Y., Takahashi M., Matsuda M., Okamoto Y., et al. Plasma concentrations of a novel, adipose‐specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000; 20: 1595–9
- Kumada M., Kihara S., Sumitsuji S., Kawamoto T., Matsumoto S., Ouchi N., et al. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol 2003; 23: 85–9
- Pischon T., Girman C. J., Hotamisligil G. S., Rifai N., Hu F. B., Rimm E. B. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004; 291: 1730–7
- Kahn R., Buse J., Ferrannini E., Stern M. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2005; 28: 2289–304
- Yarnell J. W. G., Sweetnam P. M., Marks V., Teale J. D., Bolton C. H. Insulin in ischaemic heart disease: are associations explained by triglyceride concentrations? The Caerphilly prospective study. Br Heart J 1994; 71: 293–6
- Després J. P., Lamarche B., Mauriège P., Cantin B., Dagenais G. R., Moorjani S., et al. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med 1996; 334: 952–7
- Welborn T. A., Wearne K. Coronary heart disease incidence and cardiovascular mortality in Busselton with reference to glucose and insulin concentrations. Diabetes Care 1979; 2: 154–60
- Pyörälä K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: results from two population studies in Finland. Diabetes Care 1979; 2: 131–41
- Eschwège E., Richard J. L., Thibult N., Ducimetière P., Warnet J. M., Claude J. R., et al. Coronary heart disease mortality in relation with diabetes, blood glucose and plasma insulin levels: The Paris prospective study, ten years later. Horm Metab Res 1985; 15((suppl))41–6
- Lamarche B., Tchernof A., Moorjani S., Cantin B., Dagenais G. R., Lupien P. J., et al. Small, dense low‐density lipoprotein particles as a predictor of the risk of ischemic heart disease in men. Prospective results from the Québec Cardiovascular Study. Circulation 1997; 95: 69–75
- Lamarche B., Moorjani S., Lupien P. J., Cantin B., Bernard P. M., Dagenais G. R., et al. Apolipoprotein A‐I and B levels and the risk of ischemic heart disease during a five‐year follow‐up of men in the Québec cardiovascular study. Circulation 1996; 94: 273–8
- Lemieux I., Lamarche B., Couillard C., Pascot A., Cantin B., Bergeron J., et al. Total cholesterol/HDL cholesterol ratio vs LDL cholesterol/HDL cholesterol ratio as indices of ischemic heart disease risk in men. The Quebec Cardiovascular Study. Arch Intern Med 2001; 161: 2685–92
- Pirro M., Bergeron J., Dagenais G. R., Bernard P. M., Cantin B., Després J. P., et al. Age and duration of follow‐up as modulators of the risk for ischemic heart disease associated with high plasma C‐reactive protein levels in men. Arch Intern Med 2001; 161: 2474–80
- Lemieux I., Lamarche B., St‐Pierre A., Mauriège P., Dagenais G. R., Després J. P. Evidence from the Québec Cardiovascular Study that the Framingham risk chart may not detect a significant proportion of high risk men with the features of the insulin resistance syndrome. Eur Heart J 2001; 22: 630
- Lemieux I., Pascot A., Couillard C., Lamarche B., Tchernof A., Alméras N., et al. Hypertriglyceridemic waist. A marker of the atherogenic metabolic triad (hyperinsulinemia, hyperapolipoprotein B, small, dense LDL) in men?. Circulation 2000; 102: 179–84
- Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285: 2486–97
- Balkau B., Charles M. A. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet Med 1999; 16: 442–3
- World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications. Report of a WHO Consultation, Part 1: Diagnosis and classification of Diabetes Mellitus. World Health Organization, Geneva 1999
- Alberti K. G., Zimmet P., Shaw J. The metabolic syndrome ‐ a new worldwide definition. Lancet 2005; 366: 1059–62
- Einhorn D., Reaven G. M., Cobin R. H., Ford E., Ganda O. P., Handelsman Y., et al. American College of Endocrinology position statement on the insulin resistance syndrome. Endocr Pract 2003; 9: 237–52
- Gale E. A. M. The myth of the metabolic syndrome. Diabetologia 2005; 48: 1679–83
- Tuomilehto J., Lindstrom J., Eriksson J. G., Valle T. T., Hamalainen H., Ilanne‐Parikka P., et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001; 344: 1343–50
- Knowler W. C., Barrett‐Connor E., Fowler S. E., Hamman R. F., Lachin J. M., Walker E. A., et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403
- Torgerson J. S., Hauptman J., Boldrin M. N., Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004; 27: 155–61
- Després J. P. Dyslipidaemia and obesity. Baillieres Clin Endocrinol Metab 1994; 8: 629–60
- Després J. P., Lamarche B. Effects of diet and physical activity on adiposity and body fat distribution: implications for the prevention of cardiovascular disease. Nutr Res Rev 1993; 6: 137–59
- Després J. P. Obesity and lipid metabolism: relevance of body fat distribution. Curr Opin Lipidol 1991; 2: 5–15
- Goodpaster B. H., Kelly D. E., Wing R. R., Meier A., Thaete F. L. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes 1999; 48: 839–47
- Leenen R., van der Kooy K., Deurenberg P., Seidell J. C., Weststrate J. A., Schouten F. J. M., et al. Visceral fat accumulation in obese subjects: relation to energy expenditure and response to weight loss. Am J Physiol 1992; 263: E913–9
- Zamboni M., Armellini F., Turcato E., Todesco T., Bissoli L., Bergamo‐Andreis I. A., et al. Effect of weight loss on regional body fat distribution in premenopausal women. Am J Clin Nutr 1993; 58: 29–34
- van der Kooy K., Leenen R., Seidell J. C., Deurenberg P., Droop A., Bakker C. J. Waist‐hip ratio is a poor predictor of changes in visceral fat. Am J Clin Nutr 1993; 57: 327–33
- Ross R., Dagnone D., Jones P. J., Smith H., Paddags A., Hudson R., et al. Reduction in obesity and related comorbid conditions after diet‐induced weight loss or exercise‐induced weight loss in men. A randomized, controlled trial. Ann Intern Med 2000; 133: 92–103
- Rice B., Janssen I., Hudson R., Ross R. Effects of aerobic or resistance exercise and/or diet on glucose tolerance and plasma insulin levels in obese men. Diabetes Care 1999; 22: 684–91
- Després J. P., Lemieux I., Prud'homme D. Treatment of obesity: need to focus on high risk abdominally obese patients. BMJ 2001; 322: 716–20