Abstract
The RET proto‐oncogene is the major gene involved in the pathogenesis of Hirschsprung (HSCR), a complex genetic disease characterized by lack of ganglia along variable lengths of the gut. Here we present a survey of the different molecular mechanisms through which RET mutations lead to the disease development. Among these, loss of function, gain of function, apoptosis, aberrant splicing and decreased gene expression are exemplified and considered with respect to their pathogenetic impact. In particular, RET transcription regulation represents a new insight into the outline of HSCR susceptibility, and having reached important progress in the last few years, deserves to be reviewed. Notably, gene expression impairment seems to be at the basis of the association of HSCR disease with several RET polymorphisms, allowing us to define a predisposing haplotype spanning from the promoter to exon 2. Putative functional variants, in the promoter and in intron 1, and proposed as low penetrant predisposing alleles, are presented and discussed. Finally, based on the RET mutation effects thus summarized, we attempt to derive conclusions which may be useful for HSCR risk prediction and genetic counselling.
Introduction
The human enteric nervous system (ENS) represents a considerable part of the peripheral nervous system, being characterized by a large network of neuronal interconnections necessary and sufficient to sustain the autonomous intestinal functions in the digestive process. The ENS is composed of two rings of ganglia distributed throughout the gut wall, the myenteric or Auerbach plexus (outer layer) and the submucosal or Meissner plexus (inner layer), derived from the neural crest (NC), a transient multipotent embryonic structure that originates from the neural tube. In particular, specific NC cells migrate along the primitive intestine in a cranio‐caudal direction during the 5th to 12th week of gestation. Absence of ganglion cells in both plexuses, along variable lengths of the distal intestine, is a constant finding in patients affected with Hirschsprung disease (HSCR), a congenital malformation characterized by intestinal obstruction and colonic distension in newborns, and constipation in adults, that occurs in 1 in 5000 live births Citation1. The length of aganglionosis is variable and correlates with the severity of the disease, starting from the rectum and extending up to the recto‐sigmoid junction (Short‐ or S‐form aganglionosis, 80% of cases) or beyond (Long‐ or L‐forms, 20%), these latter forms including aganglionosis to splenic flexure (colonic forms, 9%) as far as to the whole colon (total colonic aganglionosis, TCA, 3%–8%), or to the whole bowel (total intestinal).
The RET proto‐oncogene
HSCR disease is regarded as a complex genetic disease, presenting with distorted sex ratio (M:F = 4:1), incomplete penetrance and variable expressivity. Risk to siblings is highly variable, depending on gender, co‐occurrence of non‐enteric phenotypes and segment length. In particular, a dominant or additive model of heredity has been proposed for L‐forms and a recessive or multifactorial pattern for S‐forms. In addition, 80% of cases are sporadic, and 20% of patients show association with additional symptoms, with special regard to Down syndrome Citation2.
The RET proto‐oncogene was identified as the major gene in HSCR following the observation of a de novo interstitial deletion of chromosome 10 (46, XX, del10q11.21) in a patient with total colonic aganglionosis Citation3, and of two other interstitial deletions showing overall a smallest region of overlap (SRO) of less than 250 kb. In this interval RET was the only known and already cloned gene Citation4,5. In addition, RET had already proven to be associated with MEN2 syndromes, that occasionally presented co‐occurrence with HSCR disease Citation6,7.
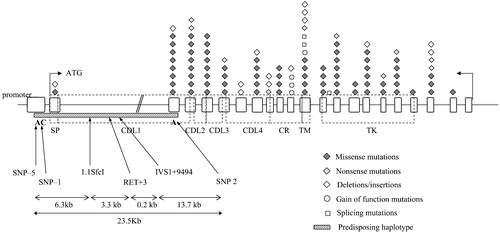
The RET proto‐oncogene, first isolated during a transfection assay (REarranged during Transfection) as a chimeric protein presenting the tyrosin‐kinase (TK) portion of RET rearranged with the 5' regions of a different gene Citation8, encodes a transmembrane TK receptor that, during development of specific neuronal cell lineages, transduces extracellular signals for cell growth and differentiation. The RET gene is composed of 21 exons scattered in a region of almost 55Kb in 10q11.2 Citation9 (). A complex pattern of alternative splicing involving the last three exons [19, 20 and 21] of the gene leads to the production of two major protein isoforms, RET9 (1072 amino acids) and RET51 (1114 amino acids) differing only for the carbossiterminal aminoacids Citation10. Several evidences suggest that the two isoforms have different biological properties and probably act as distinct signalling pathways Citation11,12. The extracellular portion of the RET protein contains 4 tandemly repeated cadherin‐like domains, each constituted of 110 residues Citation13, suggesting a role for RET also in cell‐cell interaction. The unique cysteine‐rich tract, adjacent to the transmembrane domain, is believed to play a role in folding and receptor dimerization. Finally, the intracellular portion is characterized by a bipartite TK domain, responsible for signal transduction and the subsequent cascade of events ().
Figure 1. Distribution ofRET mutations along the 21 exons. Missense, nonsense and deletion/insertion mutations conferring loss of function, together with gain‐of‐function and splicing mutations are reported using different symbols. The predisposing haplotype and relative position of its specific markers are indicated by a striped rectangle. Protein domains are reported as well. SP = signal peptide; CDL = cadherin‐like domain; CR = cysteine‐rich domain; TM = transmembrane; TK = tyrosine kinase domain.
RET activation is dependent on binding a ligand complex including members of the glial cell line‐derived neurotrophic factor (GDNF) family such as GDNF, neurturin (NTN), artemin (ART) and persepherin (PSP) and the corresponding co‐receptors, four glycosylphosphatidylinositol (GPI)‐linked proteins known as GFRα1‐>4 Citation14. In particular, the complex GDNF‐GFRα1 is considered the most relevant activator of the RET receptor. Such a binding leads to homodimerization and activation of RET, followed by auto‐phosphorylation of tyrosine residues of the RET intracellular domain. Four of these residues (Y905, Y1015, Y1062 and Y1096, this latter peculiar to the RET51 isoform) are docking sites for intracytoplasmatic effectors, such as plc‐γ, enigma and shc, activating the downstream signalling pathway. In particular, two cascades of events have been described, one involving grb2‐mSOS and leading to Ras/ERK and MAPK activation and the other occurring through the recruitment of grb2/gab2 and leading to the PI3K and Akt activation Citation15.
Key messages
Hirschsprung (HSCR) disease is regarded as a complex genetic disease. HSCR pathogenesis is sustained by loss‐of‐function effects of RET mutations
Almost 15% of patients affected by a genetic disorder might carry splicing alterations in disease‐causing genes. Common variants of conserved gene regions may be involved in expression regulation
The full comprehension of the complex inheritance of HSCR represents the first step to reliable risk prediction
RET and HSCR: Loss‐of‐function mutations
Mutations of the RET proto‐oncogene have been detected in up to 50% of familial patients and in 7%–35% of sporadic HSCR cases, depending on series Citation16–18. Nucleotide changes include microdeletions, insertions, variants affecting the correct RNA splicing, nonsense mutations, and, above all, missense mutations, with more than 100 different mutations described so far Citation19 ().
The RET mutations found in HSCR patients are scattered throughout the entire gene, do not reveal any evident genotype‐phenotype correlation and are more frequent in TCA and L‐ than in S‐forms, overall suggesting the possible effect of multiple genes which would especially work in the least severe cases. A very large proportion of mutations, especially in long aganglionosis, are de novoCitation16–18 possibly because the most severe cases have been fatal until recently, preventing the gathering of familial cases.
HSCR biology is sustained by loss‐of‐function effects of the RET mutations, resulting in abrogation of the functional gene product, a conclusion sustained by the detection of interstitial deletions of chromosome 10q11.2 associated with HSCR Citation3,Citation5, the description of RET nonsense mutations resulting in predicted non‐functional protein and the observation of missense mutations often involving amino acids conserved during evolution. However, different molecular mechanisms have also been proposed for RET mutations, like haploinsufficiency, suggested by gene deletions, and dominant negative effects, as most HSCR patients retain one functional RET allele.
Based on their location in the protein, mutations affecting the intracytoplasmatic domain could impair the kinase activity required for proper signal transduction, altering either the catalytic function, the stability of the enzyme structure or the binding of transduction effectors (). Pasini et al. demonstrated in 2005 that some intracytoplasmatic mutations abrogated the cell transforming ability when introduced into a rearranged oncogenic form of RET (RET/PTC), maybe acting through a dominant negative effect due to heterodimerization of wild‐type with mutated proteins or to competition for downstream substrates Citation20. The same conclusions were also achieved through the use of RET constructs carrying constitutively activating mutations typical of MEN2A Citation21,22. On the contrary, other intracellular RET mutations have been demonstrated to interfere with one of the RET signalling pathways required for mitogenesis. As an example, a mutation affecting residue 907 (K907E) Citation22 was postulated to disturb the nearby putative docking site located in tyr905. Similarly, two mutations, del1059 and L1061P, described in the proximity of residue Y1062, a multifunctional docking site essential for the recognition of RET by the PTB domain of Shc, were proven to abolish or partially inhibit the binding of Shc to RET Citation23.
Figure 2. Pathogenetic mechanism of different RET mutations in enteric nervous system(ENS) development. The RET signalling complex is formed by a RET dimer, the ligand GDNF and co‐receptor GRFα1 (A). Intracytoplasmatic RET mutations (B) can lead to loss of activity of the RET protein, while in presence of extracellular mutations (C) either lack of binding activity or lack of exposure to the cell surface due to misfolding can occur. Mutations of cysteine residues of the cysteine‐rich domain confer a gain of function in thyroid tissues (represented in the boxes) leading to tumour. Depending on cysteine codons involved in the substitutions, either normal (D) or impaired (E) ENS development is achieved.
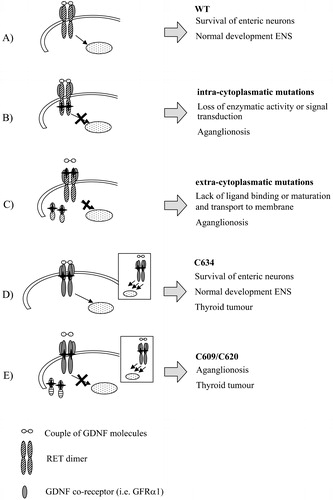
Mutations of the extracellular domain (ECD) can affect RET function through a number of different mechanisms (). Asai et al. (1995) demonstrated that mutagenesis of a putative Ca(2+)‐binding site of the cadherin‐like domain drastically decreased the MEN2A mutant constructs transforming activity. Moreover, the 170‐kDa mature RET protein was very little expressed, while expression of the 150‐kDa, known to be an immature form of RET protein retained in the endoplasmic reticulum (ER), was not altered, thus demonstrating that transport of the RET protein to the plasma membrane is essential for MEN2A mutant transforming activity Citation24. A similar result was obtained, using MEN2B mutant constructs, for another mutation affecting one of the four residues of the motif Leu‐Asp‐Arg‐Glu (LDRE), that is involved in calcium binding and highly conserved in most cadherins Citation21. Inhibition of the transport of the RET protein to the plasma membrane, possibly through impairment of functional folding, was shown also for specific HSCR mutations affecting the extracellular region of RET and not part of the cadherin motif Citation25. Either inhibition of the kinase acitivity, inability of ligand binding and/or prevention of receptor maturation and its exposure at the cell surface are all possible molecular loss‐of‐function mechanisms consistent with the haploinsufficiency model already proposed for RET in HSCR: indeed, an appropriate level of RET expression at the cell surface is required for migration of enteric neuroblasts along the gut Citation25. Finally, Cosma et al. (1998) proposed that RET mutations of the extracellular domain, resulting in the lack of the 170kDa mature form and in the retention of the 150kDa immature form in the ER, were defective in the glycosylation. Moreover, consistently with a dominant negative mechanism, the 150kDa RET species retained in the ER formed dimers and aggregates which further prevented protein maturation and function, through formation of heteromeric complex Citation26.
Mutations of the ECD have been classified into four classes, according to established or predicted effects on different functions: N‐glycosilation (I), Ca2+ coordination (II), disulfide bond formation (III) and no predictable effect (IV) Citation27. Analysis of these mutations resulted in the impairment of transport to ER and golgi and protein maturation, effects which were proven to be reversed under conditions known to facilitate protein folding. Interestingly, also a fraction of newly synthetized RET was normally recognized as misfolded and degraded by the proteasome, suggesting an intrinsic susceptibility to misfolding of the wild‐type RET molecule. In this light, the suboptimal expression of mature human RET ECD may be the result of a trade‐off meant to avoid an increased incidence of thyroid tumours, at the expense of a greater vulnerability to HSCR Citation27.
Apoptosis
A role of RET as inducer of apoptosis in the absence of its ligand GDNF, while RET pro‐apoptotic activity is blocked by GDNF, has been described Citation28. In particular, such a RET activity was shown to be caspase dependent, and the fragment lying between positions D707 and D1017, mapped as the cleavage sites for caspase, was identified as the pro‐apoptotic domain of RET. As a consequence, mutations D707N and D1017N fail to induce cell death. In accordance with these observations several HSCR intra‐ and extra‐cytoplasmatic mutations were described to be able to transform RET into a constitutive inducer of cell death both in the presence and in the absence of GDNF and independently of its kinase activity. Cell death block could be due to ligand‐dependent dimerization of the receptor and/or recruitment of proteins possibly interfering with caspase cleavage. Therefore, HSCR mutations would act by preventing, in a dominant‐negative fashion, the ligand‐binding suppression of RET pro‐apoptotic activity, thus leading to embryonic death of RET‐expressing enteric neuroblasts. Though the question whether RET plays an active or simply a passive role in apoptosis induction is still open, such a model intriguingly hypothesizes a ligand‐dependent control of the migration of RET expressing NC cells during development, not only at a guidance, but also at a survival level, and that, in HSCR, the loss of neural crest derived ganglia of the hindgut might result from RET‐induced cell death.
RET gain‐of‐function mutations and HSCR disease
Germline mutations of the RET proto‐oncogene have been found to predispose not only to HSCR disease but also to the development of other autosomal dominantly inherited disorders such as the multiple endocrine neoplasia type 2A and 2B (MEN 2A and MEN 2B), and familial medullary thyroid carcinoma (FMTC) Citation6,7. MEN 2A is characterized by the occurrence of medullary thyroid carcinoma (MTC) with variable expression of phaeochromocytoma and hyperparathyroidism. Single base‐pair substitutions in one of five codons, 609, 611, 618, 620 (exon 10) or 634 (exon 11), are found in >98% of MEN 2A families Citation29. As a result of these mutations, a cysteine residue normally involved in the intramolecular disulphide bonds that determine the tertiary structure of RET is unpaired and can form intermolecular bonds with other RET molecules, thus leading to dimerisation of the receptor and ligand‐independent activation of its TK domain Citation30. In this light, the presence of the HSCR phenotype in rare MEN2A families with specific gain‐of‐function Cys mutations (at positions 609, 618, 620 but not 634) () cannot be easily reconciled. Takahashi et al. (1999) found that cys mutations at residues 609, 618, or 620 are not only associated with constitutively RET activation but also with strong reduction of the RET amount at the plasma membrane compared to RET bearing a Cys634 mutation Citation31 (). Even low levels of RET covalent dimers resulting from mutations at Cys609, 618 or 620 are sufficient to trigger the uncontrolled proliferation of thyroid C‐cell and chromaffin cell, leading to the generation of endocrine tumours. On the other hand, such low amounts cannot compensate for the overall decrease of RET expressed at the cell surface which would position below the threshold required to elicit a trophic response in the precursors of enteric neurons, thereby leading to HSCR. Differently, the Cys634 signal would remain above the threshold, inducing a normal development of the enteric nervous system. A similar explanation has been formulated by Mograbi et al. (2001) comparing in vitro the two Cys609 and Cys620 mutants with the Cys634 RET variant for their ability to rescue neuroectodermic cells (SK‐N‐MC cells) from apoptosis. They demonstrated that like the wild‐type RET, the Cys634 RET variant can trigger antiapoptotic pathways only in response to GDNF. In contrast, Cys609 or Cys620 mutations, which impair the terminal RET glycosylation required for its insertion at the plasma membrane, abrogate GDNF‐induced cell rescue Citation32. Taken together, these data support the idea that also ligand responsiveness, and not only a proper amount of receptor at the cell surface, is mandatory to rescue neuroectodermic cells from apoptosis. Similarly, Arighi et al. (2004), demonstrated that despite Cys 620 mutation promoting high cell proliferation, the C620R‐MDCK cells are completely unable to migrate and branch in response to GDNF and are also prone to apoptosis, thus showing features common to classical loss‐of‐function HSCR‐associated mutations of RETCitation33.
So, the apparent paradox of the HSCR phenotype in combination with symptoms due to increased activity of the RET gene can be explained with the activation of different cellular programs during the differentiation of neural crest‐derived cells. In presence of Cys 609, 618, 620 mutations, insufficient RET protein in the developing enteric nervous system predisposes to HSCR, while at the same time activation of the RET signalling pathways in the thyroid/adrenal glands is sufficient to induce hyperplasia and tumour formation (). However, since HSCR and MEN2a are associated in only 25%–40% of families with a mutation in the above codons, other genetic or environmental factors must influence the penetrance of the enteric phenotypes Citation34.
Other RET mediated pathogenetic mechanisms
Mutations described above are clearly causative. Still, the mutation detection rate is lower than 50% in familial and 7%–35% in sporadic cases, so that the majority of HSCR patients still await a clarification of the underlying pathogenetic mechanism. The observation that 90% of HSCR families are linked to RET, also in the absence of clearly causative mutations, suggests a RET‐mediated pathogenesis alternative to the classical ‘coding mutation‐disease’ model. Possible explanations involve neutral gene variants acting as low penetrant alleles, or mutations in RET non‐coding regions having regulatory functions or affecting transcript processing and maturation. An example of these latter molecular pathogenetic mechanisms is given by mutations affecting RNA splicing.
Synonymous and intronic RET mutations affecting RNA splicing in HSCR disease
It has been suggested that almost 15% of patients affected by a genetic disorder might be explained by splicing alterations in disease‐causing genes. Most of these mutations directly affect the canonical consensus sequences that define exon‐intron boundaries, while others involve mutations inactivating exon splicing enhancers Citation35. Since 1994, several intronic mutations of the RET gene have been reported as putative splicing mutations in HSCR patients, but no functional test has been provided for the great majority of them, with the exception of the IVS12+19C>T for which a causative effect on HSCR development was demonstrated Citation36. RET‐silent mutations have been involved in HSCR pathogenesis too. In particular, the synonymous nucleotide change c1941C>T(I647I) was demonstrated to lead to aberrant activation of cryptic AG‐acceptor intronic sites Citation37. Similarly, the RET sequence alteration c.1947G>A(S649S) was reported to be associated with aberrant splicing of exon 11, due to creation of a sequence closely resembling the consensus splice acceptor site [(ct)10ncagG] Citation38 (). Therefore, splicing mutations can be considered as weak HSCR‐associated RET alleles which compromise but do not abrogate RET activity, thus likely requiring additional genetic changes for HSCR development.
RET expression
Common variants of conserved gene regions are often found in association with diseases, thus suggesting their involvement, as low penetrant causative alleles, in expression regulation. Alternatively, these variants may be in linkage disequilibrium (LD) (non‐random assortment of alleles at loci) with the functional causative mutation. According to such a view, several SNPs (single nucleotide polymophisms) Citation39–41 and haplotypes (combinations of alleles on a single chromosome) Citation42–45 of the RET gene have been described as under‐ or over‐represented in HSCR patients, compared to healthy controls. In particular, on the basis of association and LD analysis of an exon 2 SNP (SNP2) and other polymorphisms in the coding region of RET, the existence of a susceptibility variant in the large RET intron 1, in LD with an ancient founding locus located 20–30 kb upstream of SNP2, was suggested Citation46. Accordingly, two novel SNPs in the promoter, at –5 and –1 nucleotides from the transcription start codon were identified, in LD with SNP2. A three‐loci haplotype, named ‘ACA’, including the A variant allele of SNP2 and the A and C alleles of the promoter SNPs, and encompassing intron 1, could therefore be reconstructed Citation45 (). Association of this haplotype with HSCR has been independently confirmed in other populations Citation44,Citation47, and a recessive model of inheritance repeatedly proposed. The direct role of the A‐C‐A variants in determining lower RET expression has been debated for long, with different groups reporting discordant evidences Citation44,Citation48. Fitze et al. (2003) reported that the HSCR associated AC combination induced a lower activation of a reporter gene compared to other RET promoter SNPs haplotypes, even though a physical interaction between nuclear factors and the promoter SNPs region could not be detected by mobility shift assay Citation44. In contrast, Griseri et al. (2005) using a similar approach in different cell lines, concluded that the promoter HSCR associated alleles did not have a direct effect on RET expression. Given both the strong LD between promoter and exon 2 SNPs (23,5 kb apart) Citation44,Citation48, and the observation of low in vivo RET gene expression associated with the whole ACA haplotype, the alternative hypothesis that the ‐5A/‐1C allelic combination may represent a marker for another functional variant lying within this haplotype was advanced Citation48. Conversely, Garcia‐Barcelo et al. (2005) reported differences in expression levels among the four promoter haplotypes constructs, not relevant when using the RET basal promoter of 373 bp but significant in the context of a full length promoter (3725 bp), suggesting that the promoter SNPs could interact with additional distal regulatory elements. In addition, they showed that the promoter SNPs region interacts with the transcription factor TTF‐1 (thyroid transcription factor‐1) to activate the RET transcription, and that such effect is decreased depending on the presence of different RET promoter alleles Citation47. However, since the HSCR‐associated AC haplotype did not result in inducing the lowest expression level, the possible presence of additional functional variants in the predisposing haplotype was confirmed.
In the meanwhile, given the increased allele sharing at markers in the 5' region of the RET locus, the existence of a common ancestral RET mutation located in intron 1 and in LD with the ACA predisposing haplotype has been investigated as well. Comparative genomics combined with genetic association studies Citation49,50 have allowed to focus on a restricted number of candidate SNPs. In particular, among 86 sequence differences observed comparing the 33Kb surrounding the intron 1 in a patient homozygous for the common risk haplotype and in a control individual homozygous for the most common non‐risk haplotype, six, highly conserved in different vertebrates and localized within putative transcription factor binding sites, showed the strongest disease association and the largest distortion in allele transmission. Among these, SNP IVS1+9494C>A was the only one located in a region also conserved in a non‐mammalian species and therefore the most likely candidate HSCR‐associated variant Citation49 ().
More recently, Emison et al. (2005) have proposed a second intron 1 common variant (called RET+3) as a HSCR low penetrant mutation (). According to these authors: 1) it showed the highest association with HSCR among several markers in a 350 Kb region surrounding RET, 2) it could explain the different genetic effects in HSCR males and females, and 3) it accounted for a 10‐ to 20‐fold greater contribution to disease susceptibility than all the other known RET mutations Citation50. Notwithstanding the complete LD between RET+3 and IVS1+9494C>A SNPs, only the RET+3 over‐transmitted allele to HSCR sibs was the derived polymorphic and not the ancestral allele, an observation which prompted the authors to consider that variant as the putative functional one. The region encompassing RET+3 was demonstrated to act as a cell‐dependent expression enhancer in a mouse neuroblastoma cell line, with the HSCR associated allele significantly reducing the enhancer activity, but the same authors could not confirm such a result in a different cell line Citation50. The transcription enhancing properties of the same region have also been investigated by another group which, however, under very similar conditions, could not replicate the former observations about a role of this RET intron 1 region in increasing reporter gene expression Citation50. In conclusion, in vitro functional findings, univocally pointing to either a specific intron 1 variant, the promoter SNPs or their synergic effect as responsible for a relatively weak decrease in RET expression induction, are not yet conclusive and therefore in need of further investigation.
Conclusions
Notwithstanding the crucial role played by RET in HSCR pathogenesis, several aspects of its involvement still need to be clarified. While RET mutations are both necessary and sufficient for MEN2, they are not sufficient for HSCR: several HSCR cases seem to result from concomitant mutations of several still unidentified genes or from non‐coding RET variants, including common disease susceptibility alleles. According to such a complex model of inheritance, to poor genotype‐phenotype correlation and to low RET mutation rate in HSCR patients, mutation screening benefits appear limited. An exception may be represented by the identification of de novo mutations, mostly found in L‐HSCR cases (the most severe cases), which change the recurrent risk dramatically. Due to reduced penetrance and variable expressivity, prediction of HSCR recurrence risk remains inappropriate, often even in families where a RET mutation is known. For all these reasons, genetic counselling in HSCR families has usually a limited suitability, suggesting also against prenatal diagnosis, with the exception of systematic testing of exons 10 and 11, to exclude mutations predisposing to MEN2A, for which a prophylactic thyroidectomy would be advisable. Nevertheless, relative risk figures (2% to 33%) can be provided by taking into account the sex and length of the aganglionic segment in the proband and the gender of the sib, with the highest recurrence risk being for a male sib of a female proband affected with L‐HSCR Citation2. Also the association with other congenital anomalies should be taken into account, since its severity influences the long‐term prognosis Citation1. Moreover, it has been recently suggested that comparative RET vertebrates sequence analyses, evaluated in the context of the physico‐chemical properties of nucleotide variations, are highly predictive of their impact on protein function and disease severity, and that variants distributions in mutational classes based on gene location are predictive of clinical presentation and/or severity Citation19. However, while such prediction proved to be highly informative in the case of RET mutations in MEN2, it did not reach statistical significance for isolated HSCR, reasonably because of its complex inheritance, which implies that the disease depends neither on amino acid substitutions alone nor on a single gene. The disclosure of additional pathogenetic effects of RET, especially associated with non coding variants, will allow the full comprehension of the complex inheritance of HSCR, a first step to reliable risk prediction and genetic counselling.
References
- Chakravarti A., Lyonnet S. Hirschsprung Disease. The Metabolic & Molecular Bases of Inherited Disease, Eighth International Edition, C. R Scriver, A. L Beaudet, D Valle, W. S Sly, B Childs, K. W Kinzler, editors, et al. McGraw‐Hill, New York 2001; vol. IV: p 6231–55, In
- Badner J. A., Sieber W. K., Garver K. L., Chakravarti A. A genetic study of Hirschsprung disease. Am J Hum Genet 1990; 46: 568–80
- Martucciello G., Bicocchi M. P., Dodero P., Lerone M., Cirillo M. S., Puliti A., et al. Total colonic aganglionosis associated with interstitial deletion of the long arm of chromosome 10. Pediatr Surg Int 1992; 7: 308–10
- Luo Y., Ceccherini I., Pasini B., Matera I., Bicocchi M. P., Barone V., et al. Close linkage with the RET protooncogene and boundaries of deletion mutations in autosomal dominant Hirschsprung disease. Hum Mol Genet 1993; 2: 1803–08
- Fewtrell M. S., Tam P. K., Thomson A. H., Fitchett M., Currie J., Huson S. M., et al. Hirschsprung's disease associated with a deletion of chromosome 10 (q11.2q21.2): a further link with the neurocristopathies?. J Med Genet 1994; 31: 325–7
- Mulligan L. M., Kwok J. B., Healey C. S., Elsdon M. J., Eng C., Gardner E., et al. Germ‐line mutations of the RET proto‐oncogene in multiple endocrine neoplasia type 2A. Nature 1993; 363: 458–60
- Donis‐Keller H., Dou S., Chi D., Carlson K. M., Toshima K., Lairmore T. C., et al. Mutations in the RET proto‐oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993; 2: 851–6
- Takahashi M., Ritz J., Cooper G. M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985; 42: 581–8
- Ceccherini I., Bocciardi R., Luo Y., Pasini B., Hofstra R., Takahashi M., et al. Exon structure and flanking intronic sequences of the human RET proto‐oncogene. Biochem Biophys Res Commun 1993; 196: 1288–95
- Myers S. M., Eng C., Ponder B. A., Mulligan L. M. Characterization of RET proto‐oncogene 3' splicing variants and polyadenylation sites: a novel C‐terminus for RET. Oncogene 1995; 11: 2039–45
- De Graaf E., Sriniva S., Kilknny C., D'Agati V., Mankoo B., Costantini F., et al. Differential activities of the RET tyrosine ‐kinase recptor isoforms during mammmalian embryogenesis. Genes Dev 2001; 15: 2433–44
- Scott R. P., Eketjall S., Aineskog H., Ibanez C. F. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem 2005; 280: 13442–9
- Anders J., Kjar S., Ibanez C. F. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin‐like domains and a calcium‐binding site. J Biol Chem 2001; 276: 35808–17
- Airaksinen M. S., Titievsky A., Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant?. Mol Cell Neurosci 1999; 13: 313–25, Review
- Arighi E., Borrello M. G., Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 2005; 16: 441–67
- Angrist M., Bolk S., Thiel B., Puffenberger E. G., Hofstra R. M., Buys C. H., et al. Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet 1995; 4: 821–30
- Attie T., Pelet A., Edery P., Eng C., Mulligan L. M., Amiel J., et al. Diversity of RET proto‐oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet 1995; 4: 1381–6
- Seri M., Yin L., Barone V., Bolino A., Celli I., Bocciardi R., et al. Frequency of RET mutations in long‐ and short‐segment Hirschsprung disease. Hum Mutat 1997; 9: 243–9
- Kashuk C. S., Stone E. A., Grice E. A., Portnoy M. E., Green E. D., Sidow A., et al. Phenotype‐genotype correlation in Hirschsprung disease is illuminated by comparative analysis of the RET protein sequence. Proc Natl Acad Sci U S A 2005; 102: 8949–54
- Pasini B., Borrello M. G., Greco A., Bongarzone I., Luo Y., Mondellini P., et al. Loss of function effect of RET mutations causing Hirschsprung disease. Nat Genet 1995; 10: 35–40
- Carlomagno F., De Vita G., Berlingieri M. T., de Franciscis V., Melillo R. M., Colantuoni V., et al. Molecular heterogeneity of RET loss of function in Hirschsprung's disease. EMBO J 1996; 15: 2717–25
- Pelet A., Geneste O., Edery P., Pasini A., Chappuis S., Atti T., et al. Various mechanisms cause RET‐mediated signaling defects in Hirschsprung's disease. J Clin Invest 1998; 101: 1415–23
- Geneste O., Bidaud C., De Vita G., Hofstra R. M., Tartare‐Deckert S., Buys C. H., et al. Two distinct mutations of the RET receptor causing Hirschsprung's disease impair the binding of signalling effectors to a multifunctional docking site. Hum Mol Genet 1999; 8: 1989–99
- Asai N., Iwashita T., Matsuyama M., Takahashi M. Mechanism of activation of the ret proto‐oncogene by multiple endocrine neoplasia 2A mutations. Mol Cell Biol 1995; 15: 1613–9
- Iwashita T., Murakami H., Asai N., Takahashi M. Mechanism of ret dysfunction by Hirschsprung mutations affecting its extracellular domain. Hum Mol Genet 1996; 5: 1577–80
- Cosma M. P., Cardone M., Carlomagno F., Colantuoni V. Mutations in the extracellular domain cause RET loss of function by a dominant negative mechanism. Mol Cell Biol 1998; 18: 3321–9
- Kjaer S., Ibanez C. F. Intrinsic susceptibility to misfolding of a hot‐spot for Hirschsprung disease mutations in the ectodomain of RET. Hum Mol Genet 2003; 12: 2133–44
- Bordeaux M. C., Forcet C., Granger L., Corset V., Bidaud C., Billaud M., et al. The RET proto‐oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J 2000; 19: 4056–63
- Eng C., Clayton D., Schuffenecker I., Lenoir G., Cote G., Gagel R. F., et al. The relationship between specific RET proto‐oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996; 276: 1575–9
- Carlomagno F., Salvatore G., Cirafici A. M., De Vita G., Melillo R. M., de Franciscis V., et al. The different RET‐activating capability of mutations of cysteine 620 or cysteine 634 correlates with the multiple endocrine neoplasia type 2 disease phenotype. Cancer Res 1997; 57: 391–5
- Takahashi M., Iwashita T., Santoro M., Lyonnet S., Lenoir G. M., Billaud M. Co‐segregation of MEN2 and Hirschsprung's disease: the same mutation of RET with both gain and loss‐of‐function?. Hum Mutat 1999; 13: 331–6
- Mograbi B., Bocciardi R., Bourget I., Juhel T., Farahi‐Far D., Romeo G., et al. The sensitivity of activated Cys Ret mutants to glial cell line‐derived neurotrophic factor is mandatory to rescue neuroectodermic cells from apoptosis. Mol Cell Biol 2001; 21: 6719–30
- Arighi E., Popsueva A., Degl'Innocenti D., Borrello M. G., Carniti C., Perala N. M., et al. Biological effects of the dual phenotypic Janus mutation of ret cosegregating with both multiple endocrine neoplasia type 2 and Hirschsprung's disease. Mol Endocrinol 2004; 18: 1004–17
- Decker R. A., Peacock M. L., Watson P. Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype‐phenotype correlation. Hum Mol Genet 1998; 7: 129–34
- Cartegni L., Chew S. L., Krainer A. R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3: 285–98
- Griseri P., Mishto M., Priolo M., Pesce B., Hamel B. J., Ravazzolo R., et al. An intronic nucleotide variant of the RET proto‐oncogene causes Hirschsprung disease by interfering with RNA splicing. Gene Funct Dis 2000; 5‐6: 184–8
- Auricchio A., Griseri P., Carpentieri M. L., Betsos N., Staiano A., Tozzi A., et al. Double Heterozygosity for a RET Substitution Interfering with Splicing and an EDNRB Missense Mutation in Hirschsprung Disease. Am J Hum Genet 1999; 64: 1216–21
- Bolk S., Pelet A., Hofstra R. M., Angrist M., Salomon R., Croaker D., et al. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc Natl Acad Sci U S A 2000; 97: 268–73
- Borrego S., Saez M. E., Ruiz A., Gimm O., Lopez‐Alonso M., Antinolo G., et al. Specific polymorphisms in the RET proto‐oncogene are over‐represented in patients with Hirschsprung disease and may represent loci modifying phenotypic expression. J Med Genet 1999; 36: 771–4
- Griseri P., Sancandi M., Patrone G., Bocciardi R., Hofstra R., Ravazzolo R., et al. A single‐nucleotide polymorphic variant of the RET proto‐oncogene is underrepresented in sporadic Hirschsprung disease. Eur J Hum Genet 2000; 8: 721–4
- Fitze G., Cramer J., Ziegler A., Schierz M., Schreiber M., Kuhlisch E., et al. Association between c135G/A genotype and RET proto‐oncogene germline mutations and phenotype of Hirschsprung's disease. Lancet 2002; 359: 1200–5
- Borrego S., Ruiz A., Saez M. E., Gimm O., Gao X., Lopez‐Alonso M., et al. RET genotypes comprising specific haplotypes of polymorphic variants predispose to isolated Hirschsprung disease. J Med Genet 2000; 37: 572–8
- Carrasquillo M. M., McCallion A. S., Puffenberger E. G., Kashuk C. S., Nouri N., Chakravarti A. Genome‐wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet 2002; 32: 237–44
- Fitze G., Appelt H., Konig I. R., Gorgens H., Stein U., Walther W., et al. Functional haplotypes of the RET proto‐oncogene promoter are associated with Hirschsprung disease (HSCR). Hum Mol Genet 2003; 12: 3207–14
- Sancandi M., Griseri P., Pesce B., Patrone G., Puppo F., Lerone M., et al. Single nucleotide polymorphic alleles in the 5' region of the RET proto‐oncogene define a risk haplotype in Hirschsprung's disease. J Med Genet 2003; 40: 714–8
- Borrego S., Wright F. A., Fernandez R. M., Williams N., Lopez‐Alonso M., Davuluri R., et al. A founding locus within the RET proto‐oncogene may account for a large proportion of apparently sporadic Hirschsprung disease and a subset of cases of sporadic medullary thyroid carcinoma. Am J Hum Genet 2003; 72: 88–100
- Garcia‐Barcelo M., Ganster R. W., Lui V. C., Leon T. Y., So M. T., Lau A. M., et al. TTF‐1 and RET promoter SNPs: regulation of RET transcription in Hirschsprung's disease. Hum Mol Genet 2005; 14: 191–204
- Griseri P., Bachetti T., Puppo F., Lantieri F., Ravazzolo R., Devoto M., et al. A common haplotype at the 5' end of the RET proto‐oncogene, overrepresented in Hirschsprung patients, is associated with reduced gene expression. Hum Mutat 2005; 25: 189–95
- Burzynski G. M., Nolte I. M., Bronda A., Bos K. K., Osinga J., Plaza Menacho I., et al. Identifying candidate Hirschsprung disease‐associated RET variants. Am J Hum Genet 2005; 76: 850–8
- Emison E. S., McCallion A. S., Kashuk C. S., Bush R. T., Grice E., Lin S., et al. A common sex‐dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature 2005; 434: 857–63
- Puppo F., Musso M., Pirulli D., Griseri P., Bachetti T., Crovella S., et al. Comparative genomic sequence analysis coupled to Chromatin Immunoprecipitation: a screening procedure applied to search for regulatory elements at the RET locus. Physiol Genomics 2005; 23: 269–74