Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare life‐threatening disease in which the immune system becomes overactive due to its inability to effectively respond to infections and/or shut down the immune response to such infections. The discovery of genetic defects in the secretory pathway of natural killer (NK) cells and cytotoxic T cells in some patients with this disease has raised important questions of the role of cytotoxic cells in the control of infections and in immune regulation. This review will give a brief overview of the clinical presentation and accepted treatment of HLH. Furthermore, it will give an in‐depth review into the known genetic defects and current knowledge of the pathophysiology of this disorder, and will highlight recent evidence suggesting that cytotoxic defects in CD4+ T regulatory cells may contribute to the pathogenesis of HLH.
| Abbreviations | ||
| IL | = | interleukin |
| IFN | = | interferon |
| TNF | = | tumor necrosis virus |
| ESR | = | erythrocyte sedimentation rate |
| CMV | = | cytomegalovirus |
| HHV | = | human herpes virus |
| VSV | = | Varicella zoster virus |
| TGF | = | transforming growth factor |
| GTP | = | guanosine triphosphate |
| Ras | = | rat sarcoma oncogene |
| PBMC | = | peripheral blood mononuclear cell |
| HSV | = | herpes sinplex virus |
Clinical presentation
Hemophagocytic lymphohistiocytosis (HLH) is a highly fatal disease of children with an estimated incidence of approximately 1.2 cases/million per year Citation1. The typical clinical presentation of HLH includes persistent high‐grade fever with splenomegaly, often in the setting of an acute infection Citation2–4. Other signs include lymphadenopathy, skin rash, jaundice, edema, and central nervous system symptoms. Abnormal laboratory findings include cytopenias affecting at least two lineages, liver dysfunction (e.g. hypertriglyceridemia, hypofibrinogenemia, elevated serum transaminases, hyperbilirubinemia, elevated prothrombin time, elevated partial thromboplastin time, and elevated thrombin time), hyperferritinemia, cerebral spinal fluid (CSF) pleocytosis, hyponatremia, and hypoproteinemia Citation4,5. In addition, histopathological evaluation of the bone marrow usually demonstrates significant histiocytosis with evidence of hemophagocytic activity. Hemophagocytosis can also be observed in other affected tissues, including the spleen, lymph nodes, CSF, and liver, with the latter demonstrating histological features similar to chronic hepatitis Citation2–4. One of the most consistent laboratory findings is low or absent natural killer (NK) cell cytotoxicity Citation6–8, which correlates with the known genetic defects associated with HLH (see below).
In 1991, the Histiocyte Society proposed diagnostic guidelines for HLH based on the common clinical and laboratory findings listed above Citation3. The Histiocyte Society HLH study group recently revised these guidelines in 2004 () Citation9. It is important to realize that not all diagnostic criteria noted in need to be fulfilled before the institution of life‐saving therapies since many subjects fail to meet full diagnostic criteria until late in the course of their disease Citation3,Citation9,10. Such early therapeutic interventions often need to be implemented based on strong clinical suspicion for HLH to prevent irreversible end‐organ damage.
Table I. Diagnostic Guidelines for HLH.
Key messages
Hemophagocytic lymphohistiocytosis (HLH) is a life‐threatening disorder resulting from uncontrolled immune activation.
Genetic defects in patients with HLH and related disorders consist of mutations in genes required for cytotoxic granule release or function.
Cytotoxicity is not only important for clearance of infectious organisms, but also for the regulation of the immune system.
The hemophagocytic spectrum
HLH represents the final pathologic diagnosis of a spectrum of disorders with similar clinical and pathologic findings (). Attempts have been made to categorize HLH into two different conditions, ‘primary or familial HLH’ and ‘secondary HLH.’ Historically these attempts were made to help distinguish the cases of HLH that present during infancy and carry a higher mortality, from those due to ‘other causes’ found in secondary HLH that present later in life and have a better prognosis Citation3,Citation9. Primary HLH has been used to describe patients with a known family history of HLH or documented genetic mutations that are associated with the development of HLH (see below). Secondary HLH encompasses a number of categories that include designations such as virus‐associated hemophagocytic syndrome, infection‐associated hemophagocytic syndrome, macrophage activation syndrome, and malignancy‐associated hemophagocytic syndrome. Unfortunately, distinguishing between primary and secondary HLH is nearly impossible in many circumstances for several reasons. Primary HLH can occur at any age, not just during infancy or early childhood Citation11–13. Since many of the known genetic mutations associated with the development of HLH are inherited in an autosomal recessive manner, there is often a negative family history of HLH. In addition, underlying genetic mutations account for only ∼40% of all primary HLH subjects. Finally many of the secondary causes are associated with a high mortality, and both primary HLH and secondary HLH can present in patients with a history of a preceding infection Citation2,Citation4,Citation14–16. These observations suggest the strong need for improved diagnostic criteria that help to distinguish patients based on their clinical severity and prognosis.
Table II. Spectrum of diseases characterized by hemophagocytosis.
Genetic mutations associated with HLH
Genetic analysis of several families with children presenting with HLH before six months of age demonstrated tight linkage with the chromosomal region 10q21‐22 Citation5. Stepp et al. subsequently verified that four of these patients had homozygous nonsense mutations in the perforin gene found in this chromosomal region, resulting in the lack of protein expression and NK cell cytotoxicity Citation17. Since this initial discovery seven other genetic mutations have been associated with the development of HLH in humans () (see below). Interestingly, all seven proteins are integral components of the granule exocytosis pathway, and are critical for either the functional secretion of cytotoxic granules, or for the delivery of proteases required for apoptosis of target cells.
Figure 1. Graphical representation of the genes involved in cytotoxic granule formation, secretion, and target cell death. The first step in cellular cytotoxicity is the development of functional granules containing effector molecules(e.g. perforin, granzymes) and regulatory molecules (e.g. DPP1, serglycin, and calreticulin). Once an effector cell makes a functional secretory synapse through the interactions of adhesion molecules (e.g. LFA‐1/ICAM‐1), signal transduction events (e.g. SAP/SLAM) are initiated resulting in cell polarization by polymerization of actin/microtubules towards the secretory synapse (e.g. WASp). The preformed cytotoxic granules migrate to the secretory synapse using myosin proteins (e.g. Myo5a) and are released from the actin filaments (e.g. Rab27a) at the plasma membrane. Fusion of the granules with the plasma membrane occurs (e.g. Lyst, Munc13‐4) and effector molecules are released into the synapse. Perforin then polymerizes into pore‐forming structures in the target cell membrane that ultimately allow for the entry of granzymes into the cytoplasm of the target cells through a yet undefined mechanism. The granzymes subsequently induce apoptosis of the target cell through the cleavage/activation of multiple pro‐apoptotic target molecules in the cytoplasm, mitochondria, and nucleus. LFA‐1 = lymphocyte function associated antigen 1; ICAM‐1 = intercellular adhesion molecule 1; Lyst = lysosomal trafficking regulator gene.
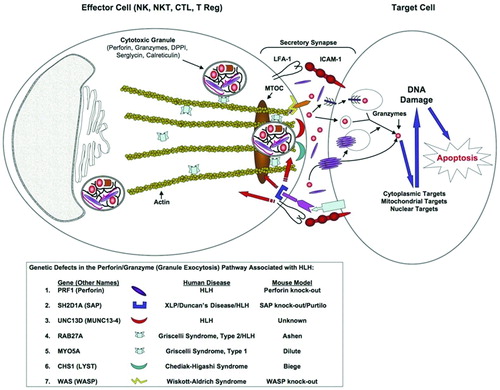
Perforin mutations in primary HLH
Perforin is a hydrophobic protein that is packaged into specialized cytotoxic granules of human natural killer (NK) cells (CD56loCD8−), natural killer T (NKT) cells (CD56loCD8+lo), and activated CD8+ T lymphocytes Citation18. Perforin is packaged with other granule regulatory proteins, including calreticulin, serglycin, and dipeptidyl peptidase I (DPPI) Citation19–21. Perforin is related to the complement component C9 Citation22, and polymerizes into a pore‐forming structure in the plasma membrane of target cells upon release by cytotoxic effector cells () Citation19,20. The critical role of perforin appears to be the introduction of cytotoxic effector molecules (e.g. granzymes) into the cytoplasm of target cells Citation21,Citation23. Granzymes are a highly conserved family of serine proteases that cleave and activate multiple cytoplasmic, mitochondrial, and nuclear targets, leading to the induction of apoptosis in the target cell Citation23,24. Perforin or granzymes alone do not appear to induce target cell death, rather the induction of target cell death requires the combination of both perforin and granzymes. How granzymes enter the cytoplasm of the target cells is controversial, and purported mechanisms include direct introduction through the perforin pore, an endocytic receptor (e.g. mannose‐6‐phosphate receptor), or some other endocytic pathway Citation19,20. Mutations in the perforin gene have been estimated to account for approximately 20%–40% percent of HLH subjects, with significant variations in the incidence of perforin gene mutations between different ethnic groups and types of HLH Citation25,26. Although the majority of patients with perforin gene mutations present with HLH in early childhood, there are rare reports of teenage and adult presentations Citation11–13,Citation27,28. To date no other cytotoxic granule protein besides perforin has been associated with the development of HLH, possibly indicating functional redundancy among the various effector granule molecules (e.g. granzymes).
UNC13D/MUNC13‐4 mutations in primary HLH
Feldman et al. demonstrated strong linkage to chromosomal region 17q25 in several HLH patients that had normal perforin expression Citation29, and showed that all of these patients had mutations in the MUNC13‐4 gene (mammalian homolog of uncoordinated, a C. elegans mutant). This gene was shown to have homology to a family of proteins found in the brain that are critical for the exocytosis of synaptic neurotransmitter vesicles. It was subsequently demonstrated that these MUNC13‐4 patient mutations failed to allow the normal release of granules from cytotoxic T cells by preventing the fusion of these cytotoxic granules to the plasma membrane () Citation29–31. Mutations in MUNC13‐4 have been found in up to 20% of primary HLH patient cohorts Citation12.
SH2D1A/SAP mutations in XLP
X‐linked lymphoproliferative (XLP) disease is an immunodeficiency that is associated with the development of lymphoma and hypogammaglobulinemia Citation32. It has been recognized that XLP patients can present in a similar fashion to HLH patients when infected with Epstein Barr virus (EBV), in that they develop life‐threatening T lymphocyte and monocyte dysregulation Citation32,33. Arico et al. subsequently demonstrated germline mutations in the XLP‐associated gene, SAP/SH2D1A (see below), in four young males with the clinical diagnosis of HLH Citation34. Two of the patients were reported to have a family history that was consistent with XLP. Based on the findings of SAP mutations in young male patients with HLH, it has been recommended that all male HLH patients undergo genetic screening for SAP mutations. Proper genetic counseling and screening should also be performed for the families of subjects identified with SAP mutations.
SAP (SLAM‐associated protein), also known as SH2D1A (src homology 2 domain protein 1A), is a small SH2 domain‐containing protein that is a critical adapter molecule between the SLAM (signaling lymphocyte activation molecule) family of immune‐cell receptors and their cognate intracellular signaling molecules () Citation35–37. Studies in SAP‐deficient mice and humans have shown that SAP is crucial for SLAM‐induced phosphorylation and recruitment of downstream signaling molecules that are important in cytokine production and cell‐mediated cytotoxicity Citation37–39.
RAB27A mutations in Griscelli's syndrome
Griscelli's syndrome (GS) is rare autosomal recessive disorder characterized by albinism and neurological defects Citation40–44. It was noted that a subset of patients with GS had variable degrees of cellular immunodeficiency and developed clinical findings consistent with HLH that can lead to death without a stem cell transplantation Citation40–44. The first gene found to be mutated in GS patients was the MYO5A gene, which encodes a myosin heavy chain that is capable of binding vesicles and moving them along actin filaments Citation45. Interestingly, the GS patients that had an associated cellular immunodeficiency were found not to have mutations in the MYO5A gene, but rather in a closely linked gene on chromosome 15q21 named RAB27ACitation46. To date, all GS patients that have been reported to develop HLH have been found to have mutations in the RAB27A gene and not the MYO5A gene Citation43,Citation46,47.
RAB27A encodes for a Ras‐like GTPase that has both Myo5a‐dependent and independent roles in the movement of vesicles (). RAB27A deficient patients (GS Type 2) have severely impaired cytotoxic activity Citation43,Citation48,49, whereas MYO5A deficient patients (GS Type 1) have normal NK and CD8 cytotoxicity Citation43,Citation48,49. GS Type 2 patients (RAB27A defects), as well as the naturally occurring mutant mouse counterpart (Ashen mice), have been shown to have a defect in the release of secretory granules from microtubules () Citation43,Citation50.
CHS1/LYST mutations in Chediak‐Higashi syndrome
Chediak‐Higashi syndrome (CHS) is a human disease associated with the clinical findings of albinism, bleeding tendency, neurological symptoms, and frequent pyogenic infections Citation51. Approximately 85% of CHS patients will enter an ‘accelerated phase’, which is manifested with symptoms that are indistinguishable from HLH patients, including hepatosplenomegaly, fever, liver dysfunction, pancytopenia, lymphadenopathy, coagulopathy, and diffuse lymphocytic and hemophagocytic infiltration of the bone marrow, liver, central nervous system, spleen, and lymph nodes Citation52,53. These patients have a substantial mortality unless they undergo stem cell transplantation Citation52,Citation54.
CHS patients display abnormally large granules in a number of cell types, including phagocytes, NK cells, and cytotoxic lymphocytes Citation51,Citation55. CHS patients have been demonstrated to have mutations in the CHS1/LYST gene (Chediak‐Higashi syndrome 1 gene/lysosomal trafficking regulator gene) Citation56,57, and, similar to the naturally occurring mouse counterpart (Beige mice), have impaired NK and T cell cytotoxicity Citation55,Citation58–60. Similar to the Munc13‐4 protein, it appears that the LYST protein is involved in the fusion of granules to the plasma membrane Citation43,Citation61.
Miscellaneous mutations associated with HLH
Wiskott‐Aldrich syndrome (WAS) is a disease that is characterized by eczema, microthrombocytopenia, and recurrent bacterial, viral and fungal infections Citation62,63. Although the clinical spectrum is heterogeneous, WAS patients typically display defects in both cellular and humoral immune responses, and are at high risk for the development of malignancies (up to 20%) and autoimmune disorders (40%–70%) Citation62,63. The WAS protein (WASp) has been shown to be an actin‐modulatory protein that is important in actin cytoskeletal reorganization after activation of various types of receptors () Citation64,65. WASp is not only crucial for cell trafficking, but also for the development of functional secretory synapses. Cells from WAS patients demonstrate defects in cell motility, homing, phagocytosis, cell adherence, and NK cell cytotoxicity. Interestingly, Pasic et al. reported a case of EBV‐associated HLH in an infant with WAS, supporting the idea that genetic mutations that fail to form a functional secretory synapse can lead to HLH Citation66.
Other immunodeficiencies that have been associated with the development of HLH include T‐B+NK+ severe combined immune deficiency (SCID) Citation67, IL‐2Rγ common chain (X‐linked T‐B+NK‐ SCID) Citation68, Omenn's syndrome (T+B‐NK+ SCID) Citation69, and DiGeorge syndrome (22q11.2 deletion) Citation70,71. These cases demonstrate the ability to develop severe HLH in the face of selective deficiencies in the cellular immune system. In addition, they underscore the importance of a thorough immunological surveillance in HLH patients, and the consideration of other immunological defects other than those that previously associated with HLH.
Pathophysiology of HLH
The pathophysiology of HLH and related disorders appears to involve the excessive activation of lymphocytes and macrophages resulting in cytokine production by these cells. Numerous cytokines are reported to be elevated in HLH, including IFN‐γ, IL‐6, IL‐10, IL‐12, IL‐16, IL‐18, and TNF‐α Citation72–76. This profound cytokine activation results in the organ dysfunction characteristic of the disorder, including hyperferritinemia and hyperlipidemia, as well as macrophage activation. The resulting organ infiltration and excessive phagocytosis in the bone marrow, liver, and other organs result in the clinical features of the syndrome. Similar observations are noted in the closely related disorder macrophage activation syndrome (MAS), which occurs in patients with systemic onset juvenile rheumatoid arthritis (SOJRA) and other autoimmune diseases. Liver histology from patients with SOJRA shows the presence of macrophages and CD8+ lymphocytes, and significant staining for TNF‐α and IFN‐γ Citation77.
The close relationship between HLH, GS, and CHS is understandable since all of the genetic mutations are involved in granule exocytosis and cytotoxicity, but why secondary HLH occurs in response to infectious organisms, malignancy, or autoimmune disease is not clear. It is possible that these associations are not coincidental, but rather represent cytotoxic defects that are yet to be characterized. Alternatively, these may represent secondary phenomenon reflecting immune dysregulation and acquired cytotoxic defects. In support of this, Fillopovich et al. studied patients with familial HLH with or without perforin defects and patients with EBV associated HLH Citation25. In familial HLH patients with or without perforin defects, they found that NK cell numbers were normal yet they had absent NK cytotoxicity, but this has not been a consistent finding in other patients with reported perforin defects Citation78. In EBV‐associated HLH, both NK numbers and cytotoxicity were reduced, suggesting that an acquired NK cell deficiency may be responsible for this clinical syndrome. Similarly, in SOJRA‐associated MAS, which is close if not identical to HLH, reports have shown defects in NK cell numbers and NK cytotoxicity Citation79. Interestingly, decreases in perforin expression have been noted in SOJRA and the levels return to normal values after autologous stem cell transplant, arguing that the defects are acquired and can be reversed Citation80. We have also observed decreased cytotoxicity in two patients (one with secondary HLH and one with SOJRA) which reversed upon treatment, also arguing that these disorders may represent acquired defects in cytotoxicity (unpublished observations).
The genetic defects in cytotoxic granule release has led to the belief that HLH is caused by the inability of cytotoxic T lymphocytes or NK cells to clear intracellular infections. Consistent with this notion, perforin deficient mice exhibit increased susceptibility to lymphocyte choriomeningitis virus (LCMV) Citation81,82, ectromelia Citation83, Herpes simplex virus Citation84, Theiler's virus Citation85, Listeria monocytogenesCitation86, Toxoplasma gondiCitation87, and Histoplasma capsulatumCitation88. These observations would argue that the failure to clear infections results in prolonged antigen presentation, excessive lymphocyte activation, and the immune pathology associated with HLH. This is supported by studies of mice exposed to LCMV showing that viral burdens were higher in perforin deficient animals and that this correlated with higher IFN‐γ levels Citation89. Furthermore, LCMV neutralizing antibodies were able to decrease the IFN‐γ levels and decrease mortality in these animals.
The failure to clear infections likely contributes to the pathology of HLH, although this cannot be the only explanation. Several studies have suggested that the lack of perforin results in a failure to regulate or terminate an immune response. Perforin deficient animals infected with LCMV displayed excessive expansion of activated CD8+ lymphocytes with significant mortality that was reversed by depletion of CD8+ cells Citation90,91. Similar studies showed that CD8+ depletion, but not NK cell or CD4+ lymphocyte depletion, rescued LCMV infected perforin deficient animals from mortality Citation89,Citation91. These studies also demonstrated that neutralizing antibodies to IFN‐γ prevented mortality, while antibodies to IL‐10, TNF‐α, IL‐12, IL‐18, macrophage colony stimulating factor, and granulocyte macrophage colony stimulating factor had no effect on mortality Citation89. Despite the increased mortality of perforin deficient mice infected with LCMV, viral titers were the same between perforin deficient and wild type animals Citation90. Additional studies have demonstrated that inoculation of perforin deficient animals with staphylococcus superantigen results in increased expansion of CD8+ cells, and peptide‐specific activation of perforin deficient CD8+ T cells results in increased numbers of CD8+ T cells Citation92. These studies show that perforin contributes to the control of CD8+ expansion independent of the failure to clear infectious organisms, and other studies support this notion. Perforin and Fas double‐deficient animals show a more aggressive autoimmune phenotype than Fas deficient animals Citation93,94. Perforin deficient animals exhibit a chronic relapsing course of experimental allergic encephalomyelitis compared with Fas deficient animals which exhibit a monophasic course Citation95. Perforin deficient animals have also been shown to exhibit exaggerated antibody responses in response to influenza antigens, again arguing that perforin may perform additional regulatory functions in immune responses Citation96.
Role of regulatory T cells in HLH
There is another possible explanation for the observed immune regulatory role of perforin/cytotoxicity not mentioned yet which involves CD4+ T regulatory cells (Tregs). Tregs have emerged as active regulators of immune responses in both humans and mice. Tregs have been grouped into two general categories, adaptive Tregs and natural Tregs, based on how they are generated, their molecular expression patterns, and their suppressive properties. Natural Tregs constitutively express the high affinity IL‐2 receptor (CD25) and the protein forkhead box P3 (FoxP3), are generated in the thymus, and suppress naïve T cell proliferation in a contact dependent manner. Adaptive Tregs are generated in the periphery to a variety of stimuli and suppress T cells responses through the secretion of immunosuppressive cytokines, such as IL‐10 and TGF‐β. When human CD4+ cells are activated with antibodies to the T cell co‐receptor (CD3) and the complement regulatory receptor CD46 (membrane cofactor protein) in the presence of IL‐2, they proliferate vigorously and express very high levels of IL‐10. Supernatants from these cells suppress the proliferation of naïve T lymphocytes in an IL‐10 dependent manner, and thus the phenotype of these cells is consistent with adaptive Tregs Citation97. These cells also express high levels of granzyme B and perforin Citation98,99. In addition, when human natural Tregs are isolated from human peripheral blood mononuclear cells (PBMCs) by flow cytometry, activated with antibodies to CD3 and CD46, and tested for granzyme expression, these cells express granzyme A but not granzyme B Citation98. When both of these cell types are used in cytotoxicity assays they exhibit killing of autologous targets, which is dependent on perforin, but independent of Fas/Fas ligand interactions. Interestingly, only activated CD4+ and CD8+ autologous targets are killed, while resting T cells are resistant to killing. Finally, CD14 positive monocytes and dendritic cells can also be killed. Importantly dendritic cells, CD8+ cells, and monocytes are the most susceptible to cytotoxicity mediated by both types of T regulatory cells, and these target cell types are central to the immune pathology of HLH Citation98,99. In addition, Janus Kinase 3 deficient animals exhibit profound myeloid expansion as they age, and these mice lack Tregs, which suggests that Tregs are necessary to prevent myeloid expansion Citation100. Furthermore, FoxP3 deficient animals, which lack natural Tregs, show an expansion of myeloid cells with myeloid infiltration of bone marrow, liver and other organs and organ pathology which is reminiscent of HLH (manuscript in preparation). No studies, however, have directly tested regulatory T cell function in patients with HLH or related disorders. Regardless, these data suggest that CD4+ lymphocytes may also exhibit perforin dependent cytotoxicity that is required for immune regulation, and the loss of this function of Tregs in perforin deficient animals and patients may contribute to the pathophysiology of HLH.
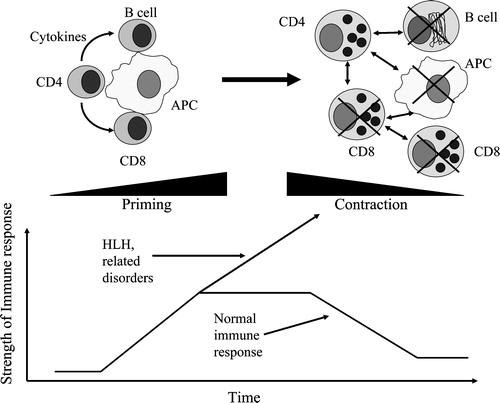
The fact that humans and mice with defects in cytotoxicity exhibit findings consistent with HLH argues strongly that cytotoxicity is important in immune regulation. We would propose a model where early in a course of an immune response, antigen presentation to CD4+ and CD8+ cells results in priming of the immune response and expansion of antigen specific cells (). These cells are then present in sufficient numbers as effector cells to eradicate viruses and other pathogens, which may be dependent on the cytotoxic function of these cells. Once the antigen load has decreased, the expanded immune system must contract to keep the final number of immune cells constant, which also appears to be dependent on functional perforin expression. Which cells are involved in the contraction phase of the immune response is not clear and may involve multiple cytotoxic cell types, such as NK cells, NKT cells, activated CD8+ cells, and/or CD4+ Tregs. The end result, however, is the effective clearance of the pathogens and contraction of the immune response. When this process is defective, the findings of excessive immune activation and HLH become apparent ().
Figure 2. Hemophagocytic lymphohistiocytosis(HLH) results from a failure of immune cell contraction due to defects in cytotoxicity. Early in the immune response, CD4 and CD8 lymphocytes are activated by antigen presenting cells and expand to be able to clear infectious pathogens. Once an effective immune response is established and the pathogens are eradicated, the expanded immune repertoire must contract to keep the relative number of lymphocytes constant. This contraction phase is dependent on cytotoxicity of lymphocytes. This may be mediated by CD8 lymphocytes, natural killer cells, or possibly by the cytotoxic T regulatory cells. Without this contraction phase, the immune response proceeds unchecked as is seen in hemophagocytic lymphohistiocytosis, resulting in the demise of the affected patients without treatment. APC: antigen presenting cell.
Therapeutic management of HLH
Untreated HLH subjects develop end‐organ damage from lymphocyte infiltration and macrophage activation. Prior to the institution of aggressive chemotherapy and immunosuppression, children with HLH had a very poor prognosis with only ∼5% of patients surviving one year from diagnosis Citation1. The first prolongation of survival was reported when these regimens were modified to include etoposide and glucocorticoids Citation101,102. Later, bone marrow transplantation became the mainstay of treatment for familial HLH, which resulted in prolonged survival and even a cure in some patients Citation103. With the introduction of the HLH‐94 protocol, which includes dexamethasone, cyclosporin A, VP‐16, and intrathecal methotrexate combined with stem cell transplantation (SCT), the three‐year overall survival rate has approached ∼50%–70% Citation104,105.
In 2004 a revised HLH treatment protocol (HLH‐2004) was developed based on the success of the HLH‐94 protocol. Similar to the HLH‐94 protocol, patients with clinical and laboratory findings consistent with HLH are aggressively treated with dexamethasone, etoposide, cyclosporin A, and in some cases intrathecal methotrexate (HLH‐2004 protocol). In addition, the primary disorder should be treated appropriately in cases of infection‐associated HLH, malignancy‐associated HLH, or other secondary causes of HLH. During this period it is important to investigate for genetic defects in perforin and SAP (if male). Assays for genetic defects in MUNC13‐4 are not yet clinically available, nor are there clinical assays for genetic defects observed with Griscelli syndrome or Chediak‐Higashi syndrome. It is important to consider functional assays of cytotoxicity (i.e. NK cytotoxicity assays) to detect defects in cytotoxicity not yet explained by available genetic tests. After eight weeks of therapy, all children with a history of familial disease, with a genetic diagnosis, or with persistent, reactivated or unresponsive disease should go immediately to allogeneic stem cell transplantation if an acceptable donor is available. Donors related to the patient should be screened for cytotoxic defects or genetic mutations to prevent transplanting a patient with a similar genetically affected bone marrow, since the timing of HLH disease presentation can vary with similar genetic mutations (see above). For those patients with secondary HLH responsive to the initial therapy, treatment should be halted to prevent the morbidity/mortality associated with stem cell transplantation. It is critical at this time, however, to closely monitor patients with secondary HLH for any signs/symptoms of relapse, and if relapse is apparent, to proceed immediately to stem cell transplantation. Potentially, cytotoxicity assays could be used to determine if the defects of cytotoxicity are reversible or persistent in patients responsive to the HLH protocol, which could help decide what type of treatment would be best suited for these patients.
Emerging therapeutic modalities
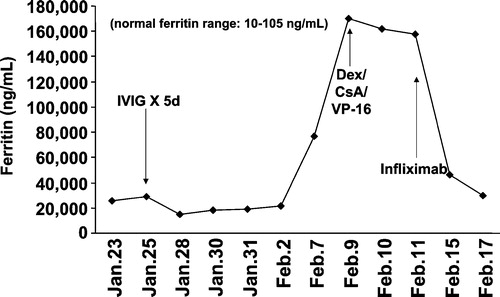
As we learn more about the pathophysiology and molecular mechanisms that lead to the clinical symptoms of HLH, additional therapeutic interventions will undoubtedly become available for treatment of patients. As noted above, two cytokines that are negatively associated with the survival of HLH subjects include high levels of IFN‐γ and TNF‐α Citation106–108, and neutralizing antibodies against IFN‐γ significantly improved the overall survival in the perforin knockout mouse model of HLH Citation89,Citation91. Given the availability of anti‐cytokine medications for humans, it would seem logical that such immune modulating drugs may be beneficial in the treatment of patients with HLH. In fact, we have treated two cases of refractory HLH with anti‐TNF‐α (Infliximab, 10 mg/kg once per week) with marked clinical and laboratory improvements () (unpublished observations, D.B. Wilson and W.J. Grossman, Washington University, 2000). In addition, there may be a role for IL‐1 neutralizing therapies in the treatment of HLH. Recently the IL‐1 receptor antagonist Anakinra has emerged as an effective therapy for SOJRA. As noted above there appear to be cytotoxic defects in patients with SOJRA, and SOJRA is associated with HLH. We have treated a patient with SOJRA and MAS with the IL‐1 receptor antagonist Anakinra with dramatic results Citation109. Although the patient was already receiving the HLH‐94 treatment protocol and was improving, it was not until Anakinra was started that his ESR, ferritin levels, and anemia normalized. He has remained symptom free, off all other therapies for almost two years on maintenance doses of Anakinra Citation109. Although this data is difficult to interpret given the multiple therapies this patient was receiving, it is a reasonable and safe treatment option to consider for patients resistant to current therapies. As new biologics become available, such as anti‐IFN‐γ, anti‐IL‐6 and anti‐IL‐18, there will certainly be other therapies in the arsenal for the treatment of HLH. There have been no studies to date which evaluate the effectiveness of these therapies, however, and these therapies should not replace the current protocols. The anecdotal evidence above was presented to raise the possibility of these therapies in the future or in patients resistant to standard therapies. The ultimate cure for patients with refractory HLH remains stem cell transplantation.
Figure 3. Effective treatment of a refractory hemophagocytic lymphohistiocytosis patient with Infliximab(anti‐TNF‐α). Briefly, the 16‐year‐old male subject had EBV‐associated hemophagocytic lymphohistiocytosis (HLH) that was refractory to IVIG, steroids, and VP‐16, leading to pulmonary failure and mechanical ventilation. Upon institution of Infliximab therapy his clinical status improved significantly within 24–48 hours, represented by a marked decrease in his serum ferritin levels and improvement of all clinical parameters including removal from mechanical ventilation.
The study of patients with HLH over the past several decades has led not only to substantial improvements in the care and survival of these patients, but also to significant new insights into the molecular defects and pathophysiology of a once fatal disease. This success story could not have been possible without the generation of an HLH registry, an active international collaborative effort, or parent organizations which helped to raise awareness and funding opportunities for this once universally fatal disease (http://histio.org/society/). With further study of the role of cytotoxicity in immune regulation, not only will the care for affected patients continue to improve, but also the scientific community will continue to gain knowledge of immune regulation, information that could prove vital to the treatment of other diseases in the future.
Acknowledgements
This work was supported by the Advancing a Healthier Wisconsin program (WJG), the Hope Street Kids (WJG), and the Abbott Scholars Award in Rheumatology Research (JWV). We thank David Wilson for his critical review of this manuscript.
References
- Henter J. I., Elinder G., Soder O., Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scan 1991; 80: 428–35
- Henter J. I., Arico M., Elinder G., Imashuku S., Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998; 12: 417–33
- Henter J. I., Elinder G., Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol 1991; 18: 29–33
- Arico M., Janka G., Fischer A., Henter J. I., Blanche S., Elinder G., et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia 1996; 10: 197–203
- Dufourcq‐Lagelouse R., Pastural E., Barrat F. J., Feldmann J., Le Deist F., Fischer A., et al. Genetic basis of hemophagocytic lymphohistiocytosis syndrome (Review). Int J Mol Med 1999; 4: 127–33
- Perez N., Virelizier J. L., Arenzana‐Seisdedos F., Fischer A., Griscelli C. Impaired natural killer activity in lymphohistiocytosis syndrome. J Pediatr 1984; 104: 569–73
- Arico M., Nespoli L., Maccario R., Montagna D., Bonetti F., Caselli D., et al. Natural cytotoxicity impairment in familial haemophagocytic lymphohistiocytosis. Arch Dis Child 1988; 63: 292–6
- Sullivan K. E., Delaat C. A., Douglas S. D., Filipovich A. H. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 1998; 44: 465–8
- Group HLS. HLH‐2004: Treatment Protocol of the Second International HLH Study 2004. 2004. (http://histio.org/society/protocols/trials‐protocols.shtml).
- Henter J. I., Elinder G. Cerebromeningeal haemophagocytic lymphohistiocytosis. [see comment]. Lancet 1992; 339: 104–7
- Allen M., De Fusco C., Legrand F., Clementi R., Conter V., Danesino C., et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be?. Haematologica 2001; 86: 499–503
- Ishii E., Ueda I., Shirakawa R., Horiuchi H., Ohga S., Furuno K., et al. Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions. Blood 2005; 105: 3442–8
- Clementi R., Emmi L., Maccario R., Liotta F., Moretta L., Danesino C., et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood 2002; 100: 2266–7
- Janka G., Imashuku S., Elinder G., Schneider M., Henter J. I. Infection‐ and malignancy‐associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am 1998; 12: 435–44
- Imashuku S., Hibi S., Tabata Y., Sako M., Sekine Y., Hirayama K., et al. Biomarker and morphological characteristics of Epstein‐Barr virus‐related hemophagocytic lymphohistiocytosis. Med Pediatr Oncol 1998; 31: 131–7
- Imashuku S., Tabata Y., Teramura T., Hibi S. Treatment strategies for Epstein‐Barr virus‐associated hemophagocytic lymphohistiocytosis (EBV‐HLH). Leuk Lymphoma 2000; 39: 37–49
- Stepp S. E., Dufourcq‐Lagelouse R., Le Deist F., Bhawan S., Certain S., Mathew P. A., et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999; 286: 1957–9
- Masson D., Tschopp J. Isolation of a lytic, pore‐forming protein (perforin) from cytolytic T‐lymphocytes. J Biol Chem 1985; 260: 9069–72
- Catalfamo M., Henkart P. A. Perforin and the granule exocytosis cytotoxicity pathway. Curr Opin Immunol 2003; 15: 522–7
- Raja S. M., Metkar S., Froehlich J. Cytotoxic granule‐mediated apoptosis: unraveling the complex mechanism. Curr Opin Immunol 2003; 15: 528–32
- Lieberman J. The ABCs of granule‐mediated cytotoxicity: new weapons in the arsenal. Nature Rev Immunol 2003; 3: 361–70
- Young J. D., Cohn Z. A., Podack E. R. The ninth component of complement and the pore‐forming protein (perforin 1) from cytotoxic T cells: structural, immunological, and functional similarities. Science 1986; 233: 184–90
- Russell J. H., Ley T. J. Lymphocyte‐mediated cytotoxicity. Annu Rev Immunol 2002; 20: 323–70
- Grossman W. J., Revell P. A., Lu Z. H., Johnson H. J., Bredemeyer A. J., Ley T. J. The orphan granzymes of humans and mice. Curr Opin Immunol 2003; 15: 544–42
- Kogawa K., Lee S. M., Villanueva J., Marmer D., Sumegi J., Filipovich A. H. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood 2002; 99: 61–6
- Ueda I., Morimoto A., Inaba T., Yagi T., Hibi S., Sugimoto T., et al. Characteristic perforin gene mutations of haemophagocytic lymphohistiocytosis patients in Japan. Br J Haematol 2003; 121: 503–10
- Ishii E., Ohga S., Tanimura M., Imashuku S., Sako M., Mizutani S., et al. Clinical and epidemiologic studies of familial hemophagocytic lymphohistiocytosis in Japan. Japan LCH Study Group. Med Pediatr Oncol 1998; 30: 276–83
- Molleran Lee S., Villanueva J., Sumegi J., Zhang K., Kogawa K., Davis J., et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet 2004; 41: 137–44
- Feldman J., Callebaut I., Raposo G., Certain S., Bacq D., Dumont C., et al. Munc13‐4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 2003; 115: 461–73
- Neeft M., Wieffer M., de Jong A. S., Negroiu G., Metz C. H., van Loon A., et al. Munc13‐4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell 2005; 16: 731–41
- Rothman J. E. Lasker Basic Medical Research Award. The machinery and principles of vesicle transport in the cell. Nat Med 2002; 8: 1059–62
- Purtilo D. T., Cassel C. K., Yang J. P., Harper R. X‐linked recessive progressive combined variable immunodeficiency (Duncan's disease). Lancet 1975; 1: 935–40
- Hamilton J. K., Paquin L. A., Sullivan J. L., Maurer H. S., Cruzi F. G., Provisor A. J., et al. X‐linked lymphoproliferative syndrome registry report. J Pediatr 1980; 96: 669–73
- Arico M., Imashuku S., Clementi R., Hibi S., Teramura T., Danesino C., et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X‐linked lymphoproliferative disease gene. Blood 2001; 97: 1131–3
- Latour S., Veillette A. Molecular and immunological basis of X‐linked lymphoproliferative disease. Immunol Rev 2003; 192: 212–24
- Latour S., Veillette A. The SAP family of adaptors in immune regulation. Semin Immunol 2004; 16: 409–19
- Veillette A., Latour S. The SLAM family of immune‐cell receptors. Curr Opin Immunol 2003; 15((3))277–85
- Sharifi R., Sinclair J. C., Gilmour K. C., Arkwright P. D., Kinnon C., Thrasher A. J., et al. SAP mediates specific cytotoxic T‐cell functions in X‐linked lymphoproliferative disease. Blood 2004; 103: 3821–7
- Gaspar H. B., Sharifi R., Gilmour K. C., Thrasher A. J. X‐linked lymphoproliferative disease: clinical, diagnostic and molecular perspective. Br J Haematol 2002; 119: 585–95
- Klein C., Philippe N., Le Deist F., Fraitag S., Prost C., Durandy A., et al. Partial albinism with immunodeficiency (Griscelli syndrome). J Pediatr 1994; 125: 886–95
- Pastural E., Ersoy F., Yalman N., Wulffraat N., Grillo E., Ozkinay F., et al. Two genes are responsible for Griscelli syndrome at the same 15q21 locus. Genomics 2000; 63: 299–306
- Trambas C. M., Griffiths G. M. Delivering the kiss of death. Nat Immunol 2003; 4: 399–403
- Stinchcombe J., Bossi G., Griffiths G. M. Linking albinism and immunity: The secrets of secretory lysosomes. Science 2004; 305: 55–59
- Blott E. J., Griffiths G. M. Secretory lysosomes. Nat Rev Mol Cell Biol 2002; 3: 122–31
- Pastural E., Barrat F. J., Dufourcq‐Lagelouse R., Certain S., Sanal O., Jabado N., et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin‐Va gene. [erratum appears in Nat Genet 1999 Nov;23(3):373]. Nat Genet 1997; 16: 289–92
- Menasche G., Pastural E., Feldmann J., Certain S., Ersoy F., Dupuis S., et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000; 25: 173–6
- Kumar M., Sackey K., Schmalstieg F., Trizna Z., Elghetany M. T., Alter B. P. Griscelli syndrome: rare neonatal syndrome of recurrent hemophagocytosis. J Pediatr Hematol Oncol 2001; 23: 464–8
- Stinchcombe J. C., Barral D. C., Mules E. H., Booth S., Hume A. N., Machesky L. M., et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol 2001; 152: 825–34
- Stinchcombe J. C., Page L. J., Griffiths G. M. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic 2000; 1: 435–44
- Haddad E. K., Wu X., Hammer J. A 3rd., Henkart P. A. Defective granule exocytosis in Rab27a‐deficient lymphocytes from Ashen mice. [see comment]. J Cell Biol 2001; 152: 835–42
- Blume R. S., Wolff S. M. The Chediak‐Higashi syndrome: studies in four patients and a review of the literature. Medicine 1972; 51: 247–80
- Introne W., Boissy R. E., Gahl W. A. Clinical, molecular, and cell biological aspects of Chediak‐Higashi syndrome. Mol Genet Metab 1999; 68: 283–303
- Rubin C. M., Burke B. A., McKenna R. W., McClain K. L., White J. G., Nesbit M. E., Jr, et al. The accelerated phase of Chediak‐Higashi syndrome. An expression of the virus‐associated hemophagocytic syndrome?. Cancer 1985; 56: 524–30
- Haddad E., Le Deist F., Blanche S., Benkerrou M., Rohrlich P., Vilmer E., et al. Treatment of Chediak‐Higashi syndrome by allogenic bone marrow transplantation: report of 10 cases. Blood 1995; 85: 3328–33
- Baetz K., Isaaz S., Griffiths G. M. Loss of cytotoxic T lymphocyte function in Chediak‐Higashi syndrome arises from a secretory defect that prevents lytic granule exocytosis. J Immunol 1995; 154: 6122–31
- Barbosa M. D., Nguyen Q. A., Tchernev V. T., Ashley J. A., Detter J. C., Blaydes S. M., et al. Identification of the homologous beige and Chediak‐Higashi syndrome genes. [erratum appears in Nature 1997;385:97]. Nature 1996; 382: 262–5
- Perou C. M., Moore K. J., Nagle D. L., Misumi D. J., Woolf E. A., McGrail S. H., et al. Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet 1996; 13: 303–8
- Roder J. C., Haliotis T., Laing L., Kozbor D., Rubin P., Pross H., et al. Further studies of natural killer cell function in Chediak‐Higashi patients. Immunology 1982; 46: 555–60
- Roder J. C. Characterization of a murine model (beige) for a natural killer cell immunodeficiency in the Chediak‐Higashi syndrome of man. Prog Clin Biol Res 1982; 94: 315–25
- Haliotis T., Roder J., Klein M., Ortaldo J., Fauci A. S., Herberman R. B. Chediak‐Higashi gene in humans I. Impairment of natural‐killer function. J Exp Med 1980; 151: 1039–48
- Ward D. M., Shiflett S. L., Kaplan J. Chediak‐Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med 2002; 2: 469–77
- Sullivan K. E., Mullen C. A., Blaese R. M., Winkelstein J. A. A multiinstitutional survey of the Wiskott‐Aldrich syndrome. J Pediatr 1994; 125: 876–85
- Imai K., Morio T., Zhu Y., Jin Y., Itoh S., Kajiwara M., et al. Clinical course of patients with WASP gene mutations. Blood 2004; 103: 456–64
- Burns S., Cory G. O., Vainchenker W., Thrasher A. J. Mechanisms of WASp‐mediated hematologic and immunologic disease. Blood 2004; 104: 3454–62
- Notarangelo L. D., Ochs H. D. Wiskott‐Aldrich Syndrome: a model for defective actin reorganization, cell trafficking and synapse formation. Curr Opin Immunol 2003; 15: 585–91
- Pasic S., Micic D., Kuzmanovic M. Epstein‐Barr virus‐associated haemophagocytic lymphohistiocytosis in Wiskott‐Aldrich syndrome. Acta Paediatr 2003; 92: 859–61
- Schmid I., Reiter K., Schuster F., Wintergerst U., Meilbeck R., Nicolai T., et al. Allogeneic bone marrow transplantation for active Epstein‐Barr virus‐related lymphoproliferative disease and hemophagocytic lymphohistiocytosis in an infant with severe combined immunodeficiency syndrome. Bone Marrow Transplant 2002; 29: 519–21
- Grunebaum E., Zhang J., Dadi H., Roifman C. M. Haemophagocytic lymphohistiocytosis in X‐linked severe combined immunodeficiency. Br J Haematol 2000; 108: 834–7
- Aleman K., Noordzij J. G., de Groot R., van Dongen J. J., Hartwig N. G. Reviewing Omenn syndrome. Eur J Pediatr 2001; 160: 718–25
- Arico M., Bettinelli A., Maccario R., Clementi R., Bossi G., Danesino C. Hemophagocytic lymphohistiocytosis in a patient with deletion of 22q11.2. Am J Med Genet 1999; 87: 329–30
- Touraine R. L., Pondarre C., Till M., Bertrand Y. Hemophagocytic lymphohistiocytosis and del22q11 [abstract]. Genetic Counseling 1999; 10: 114–5
- Osugi Y., Hara J., Tagawa S., Takai K., Hosoi G., Matsuda Y., et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 1997; 89: 4100–3
- Schneider E. M., Lorenz I., Muller‐Rosenberger M., Steinbach G., Kron M., Janka‐Schaub G. E. Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer‐cell‐induced apoptosis. Blood 2002; 100: 2891–8
- Takada H., Nomura A., Ohga S., Hara T. Interleukin‐18 in hemophagocytic lymphohistiocytosis. Leuk Lymphoma 2001; 42: 21–8
- Takada H., Takahata Y., Nomura A., Ohga S., Mizuno Y., Hara T. Increased serum levels of interferon‐gamma‐inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol 2003; 133: 448–53
- Takada H., Ohga S., Mizuno Y., Nomura A., Hara T. Increased IL‐16 levels in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2004; 26: 567–73
- Billiau A. D., Roskams T., Damme‐Lombaerts R., Matthys P., Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN‐gamma‐producing lymphocytes and IL‐6‐ and TNF‐alpha‐producing macrophages. Blood 2005; 105: 1648–51
- Grossman W. J., Radhi M., Schauer D., Gerday E., Grose C., Goldman F. D. Development of hemophagocytic lymphohistiocytosis in triplets infected with HHV‐8. Blood 2005; 106: 1203–06
- Grom A. A., Villanueva J., Lee S., Goldmuntz E. A., Passo M. H., Filipovich A. Natural killer cell dysfunction in patients with systemic‐onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr 2003; 142: 292–6
- Wulffraat N. M., Rijkers G. T., Elst E., Brooimans R., Kuis W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem‐cell transplantation. Rheumatology (Oxford) 2003; 42: 375–9
- Kagi D., Ledermann B., Burki K., Seiler P., Odermatt B., Olsen K. J., et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin‐deficient mice. Nature 1994; 369: 31–7
- Walsh C. M., Matloubian M., Liu C. C., Ueda R., Kurahara C. G., Christensen J. L., et al. Immune function in mice lacking the perforin gene. Proc Natl Acad Sci U S A 1994; 91: 10854–8
- Mullbacher A., Hla R. T., Museteanu C., Simon M. M. Perforin is essential for control of ectromelia virus but not related poxviruses in mice. J Virol 1999; 73: 1665–7
- Ghiasi H., Cai S., Perng G., Nesburn A. B., Wechsler S. L. Perforin pathway is essential for protection of mice against lethal ocular HSV‐1 challenge but not corneal scarring. Virus Res 1999; 65: 97–101
- Rossi C. P., McAllister A., Tanguy M., Kagi D., Brahic M. Theiler's virus infection of perforin‐deficient mice. J Virol 1998; 72: 4515–9
- Kagi D., Ledermann B., Burki K., Hengartner H., Zinkernagel R. M. CD8+ T cell‐mediated protection against an intracellular bacterium by perforin‐dependent cytotoxicity. Eur J Immunol 1994; 24: 3068–72
- Denkers E. Y., Yap G., Scharton‐Kersten T., Charest H., Butcher B. A., Caspar P., et al. Perforin‐mediated cytolysis plays a limited role in host resistance to Toxoplasma gondii. J Immunol 1997; 159: 1903–8
- Zhou P., Freidag B. L., Caldwell C. C., Seder R. A. Perforin is required for primary immunity to Histoplasma capsulatum. J Immunol 2001; 166: 1968–74
- Jordan M. B., Hildeman D., Kappler J., Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 2004; 104: 735–43
- Matloubian M., Suresh M., Glass A., Galvan M., Chow K., Whitmire J. K., et al. A role for perforin in downregulating T‐cell responses during chronic viral infection. J Virol 1999; 73: 2527–36
- Badovinac V. P., Hamilton S. E., Harty J. T. Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin‐deficient mice. Immunity 2003; 18: 463–74
- Kagi D., Odermatt B., Mak T. W. Homeostatic regulation of CD8+ T cells by perforin. Eur J Immunol 1999; 29: 3262–72
- Peng S. L., Moslehi J., Robert M. E., Craft J. Perforin protects against autoimmunity in lupus‐prone mice. J Immunol 1998; 160: 652–60
- Spielman J., Lee R. K., Podack E. R. Perforin/Fas‐ligand double deficiency is associated with macrophage expansion and severe pancreatitis. J Immunol 1998; 161: 7063–70
- Malipiero U., Frei K., Spanaus K. S., Agresti C., Lassmann H., Hahne M., et al. Myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis is chronic/relapsing in perforin knockout mice, but monophasic in Fas‐ and Fas ligand‐deficient lpr and gld mice. Eur J Immunol 1997; 27: 3151–60
- Liu B., Mori I., Hossain M. J., Dong L., Chen Z., Kimura Y. Local immune responses to influenza virus infection in mice with a targeted disruption of perforin gene. Microb Pathog 2003; 34: 161–7
- Kemper C., Chan A. C., Green J. M., Brett K. A., Murphy K. M., Atkinson J. P. Activation of human CD4+ cells with CD3 and CD46 induces a T‐regulatory cell 1 phenotype. Nature 2003; 421: 388–92
- Grossman W. J., Verbsky J. W., Barchet W., Colonna M., Atkinson J. P., Ley T. J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004; 21: 589–601
- Grossman W. J., Verbsky J. W., Tollefson B. L., Kemper C., Atkinson J. P., Ley T. J. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 2004; 104: 2840–8
- Grossman W. J., Verbsky J. W., Yang L., Berg L. J., Fields L. E., Chaplin D. D., et al. Dysregulated myelopoiesis in mice lacking Jak3. Blood 1999; 94: 932–9
- Henter J. I., Elinder G., Finkel Y., Soder O. Successful induction with chemotherapy including teniposide in familial erythrophagocytic lymphohistiocytosis. Lancet 1986; 2: 1402
- Fisher A., Virelizier J. L., Arenzana F., Perez A., Nezelof C., Griscelli C. Treatment of four patients with erythrophagocytic lymphohistiocytosis by a combination of epipodophyllotoxin, steroids, intrathecal methotrexate and cranial irradiation. Pediatrics 1985; 76: 263–8
- Bolme P., Henter J. I., Winiarski J., Elinder G., Ljungman P., Lonnerholm G., et al. Allogeneic bone marrow transplantation for hemophagocytic lymphohistiocytosis in Sweden. Bone Marrow Transplant 1995; 15: 331–5
- Baker K. S., DeLaat C. A., Steinbuch M., Gross T. G., Shapiro R. S., Loechelt B., et al. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood 1997; 89: 3857–63
- Henter J. I., Samuelsson‐Horne A., Arico M., Egeler R. M., Elinder G., Filipovich A. H., et al. Treatment of hemophagocytic lymphohistiocytosis with HLH‐94 immunochemotherapy and bone marrow transplantation. Blood 2002; 100: 2367–73
- Ishii E., Ohga S., Aoki T., Yamada S., Sako M., Tasaka H., et al. Prognosis of children with virus‐associated hemophagocytic syndrome and malignant histiocytosis: correlation with levels of serum interleukin‐1 and tumor necrosis factor. Acta Haematol 1991; 85: 93–9
- Imashuku S., Hibi S., Fujiwara F., Ikushima S., Todo S. Haemophagocytic lymphohistiocytosis, interferon‐gamma‐naemia and Epstein‐Barr virus involvement. Br J Haematol 1994; 88: 656–8
- Henter J. I., Elinder G., Soder O., Hansson M., Andersson B., Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 1991; 78: 2918–22
- Verbsky J. W., White A. J. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol 2004; 31: 2071–5
- Fisman D. N. Hemophagocytic syndromes and infection. Emerg Infect Dis 2000; 6: 601–8