Abstract
Antiplatelet therapy has been established as a preventive medicine for ischemic cardiovascular diseases both at acute and chronic phases. This therapy is also crucial for the prevention of thrombotic events after coronary stent implantation. So far, many lines of clinical evidence have demonstrated the beneficial effects of aspirin (an irreversible cyclooxygenase inhibitor) and thienopyridine derivatives (adenosine diphosphate (ADP)‐receptor P2Y12 inhibitors). Recently, it has been reported that the cardiovascular risk is elevated in patients with platelets resistant to these drugs, compared to the good responders. One of the current problems to be solved in antiplatelet therapy is to find out patients resistant to the antiplatelet therapy and improve its preventive effects. In addition to aspirin and thienopyridines, several types of drugs with antiplatelet effects are currently available in clinical practice. Clinical evidence has recently been accumulating for these drugs that can be potential alternatives in patients with aspirin or thienopyridine resistance. In this review, the mechanisms, evidence and approach to the present problems of drugs with antiplatelet effects are discussed.
| Abbreviations | ||
| 5‐HT | = | 5‐hydroxytriptamine |
| ADP | = | adenosine diphosphate |
| ATP | = | adenosine triphosphate |
| cAMP | = | cyclic adenosine monophosphate |
| cGMP | = | cyclic guanosine monophosphate |
| COX | = | cyclooxygenase |
| DHA | = | docosahexaenoic acid |
| EPA | = | eicosapentaenoic acid |
| GP | = | glycoprotein |
| HMG‐CoA | = | 3‐hydroxy‐3‐methylglutaryl coenzyme A |
| NSAID | = | non‐steroidal anti‐inflammatory drug |
| PAD | = | peripheral artery disease |
| PAR | = | protease‐activated receptor |
| PCI | = | percutaneous coronary intervention |
| PDE | = | phosphodiesterase |
| PDGF | = | platelet‐derived growth factor |
| PKA | = | cAMP‐dependent protein kinase |
| PKG | = | cGMP‐dependent protein kinase |
| PRP | = | platelet‐rich plasma |
| RR | = | relative risk |
| SNP | = | single nucleotide polymorphism |
| TF | = | tissue factor |
| VASP | = | vasodilator‐stimulated phosphoprotein |
| vWF | = | von Willebrand factor |
Introduction: Molecular mechanism of platelet activation
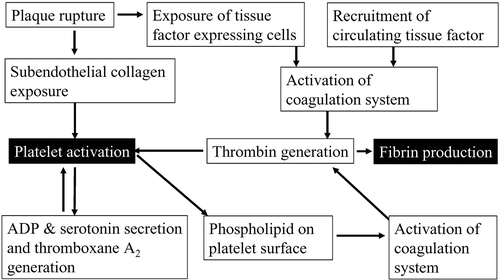
Acute coronary syndrome and cerebral infarction are caused by thrombotic occlusion of arteries. When plaque rupture or erosion occurs in coronary arteries, subendothelial tissue is exposed to the lumen Citation1–4. Collagen in this tissue recruits von Willebrand factors (vWFs) from the blood stream, and collagen‐bound vWFs change its conformation in a high‐shear condition to become competent to interact with glycoprotein (GP) Ib/V/IX complex on the platelet surface () Citation3,4. This interaction causes ‘rolling’ of platelets on the vessel wall. Subsequently, platelets adhere tightly through collagen receptors such as integrin α2β1 (GPIa/IIa) and GPVI Citation3,4. There, platelets are activated and a series of reactions including granule secretion, aggregation and shape change occur, resulting in the formation of ‘white’ thrombus (). In activated platelets, phosphatidyl serine emerges on the surface by a flip‐flop mechanism and provides a place for coagulation cascades (). Cells, such as macrophages, in subendothelial tissues of atheromatous plaque express tissue factors on their surface. Further, circulating tissue factors Citation5 possibly present on monocyte‐derived microparticles gather at the growing thrombus through the interaction with P‐selectin on the plasma membrane of activated platelets, which is translocated by α‐granule secretion Citation3,4. These tissue factors trigger the coagulation cascade to generate thrombin at the last step. The functions of thrombin, a serine protease, are not only to generate fibrin by cleaving fibrinogen, but also to activate platelets through thrombin receptors. Among four protease‐activated receptor (PAR) family members, PAR‐1 is the major thrombin receptor in human platelets Citation6. Thus, platelets initially play a central role in arterial thrombus formation and, at later stages, collaborate with the coagulation system to form ‘red’ thrombus, resulting in occlusion of arteries ().
Figure 1. Molecular mechanism of platelet activation. Activated platelets exert functions such as adhesion, granule secretion and aggregation as described in the text.
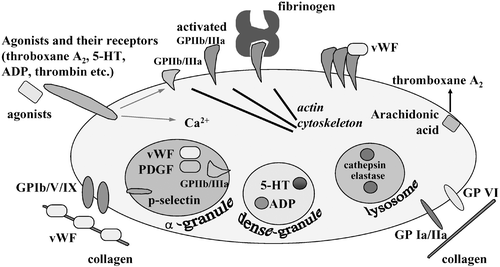
Figure 2. Synergistic interaction between platelet activation and coagulation system. The interaction of platelets and coagulation system following plaque rupture is schematically shown.
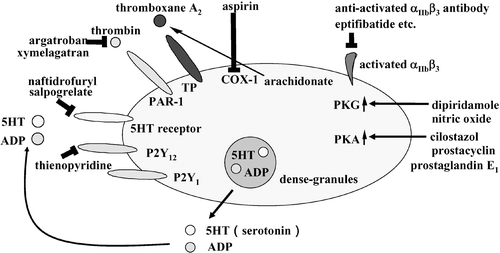
Platelets contain three types of vesicles including α‐granules, dense granules and lysosomes Citation3,4. They are secreted in response to increased intracellular calcium ion concentration in activated platelets () Citation7. α‐Granules contain protein factors such as platelet‐derived growth factor (PDGF), vWF and fibrinogen. P‐selectin and GPIIb/IIIa (integrin αIIbβ3) are membrane proteins stored on α‐granule membranes. These are translocated to the plasma membrane upon α‐granule exocytosis. Secreted hydrolytic enzymes stored in lysosomes would contribute to vascular remodeling. Dense granules contain adenosine diphosphate (ADP) and 5‐hydroxytryptamine (5‐HT or serotonin). Through receptors on platelets, ADP and 5‐HT enhance platelet activation in positive feedback manners. Activated platelets produce thromboxane A2 through the arachidonic acid cascade. Thromboxane A2 is also a self‐agonist. These agonists together with thrombin play roles in synergistic activation of platelets at the site of thrombus formation. Therefore, drugs that block one receptor pathway are used as antiplatelet drugs ().
Figure 3. Molecular targets of antiplatelet drugs. Target molecules of the drugs with antiplatelet activity are shown as described in the text.
Platelet aggregation is mediated by GPIIb/IIIa that is abundantly expressed in platelets () Citation3,4. More than half GPIIb/IIIa molecules are present on the plasma membrane and some are in α‐granules in unstimulated platelets. Although GPIIb/IIIa has no ligand‐binding activity in unstimulated platelets, it undergoes a conformational change upon platelet activation to become able to bind its ligands, vWF and fibrinogen Citation3,4. Both ligands have multiple binding sites for activated GPIIb/IIIa and thereby induce platelet aggregation by bridging adjacent platelets. Activation of this integrin is considered the ‘common final pathway’ since all the signaling pathways utilize this molecule at the last step toward aggregation.
Antiplatelet therapy has been established as a preventive medicine for cardiovascular events. So far, many lines of clinical studies have demonstrated beneficial effects in the therapy with aspirin and thienopyridine derivatives. Several types of other drugs with antiplatelet effects are also available in clinical practice and evidence has been accumulating for such drugs. Recently, it has been noticed that the cardiovascular risk is elevated in patients with platelets poorly responsive to aspirin or thienopyridine, compared to the good responders, a phenomenon called antiplatelet drug resistance. This problem should be solved by establishing a strategy to find out which patients are resistant to the therapy and to improve its preventive effects. In this review, the mechanisms, evidence and present topics of antiplatelet drugs are discussed.
Key messages
Antiplatelet therapy has been established as a preventive medicine for ischemic cardiovascular disease.
In this review, concerning antiplatelet drugs, not only aspirin and thienopyridine derivatives but also other types of drugs, the molecular mechanisms, recent evidence, and current problems such as aspirin resistance are discussed for the better antiplatelet therapy.
Drugs with antiplatelet effects
Aspirin
Mechanism and evidence
In stimulated platelets, phospholipase A2 is activated to generate arachidonic acid by cleaving phospholipids. Thromboxane A2 is produced from arachidonic acid through a series of reactions including an initial step mediated by cyclooxygenase (COX) Citation3. Thromboxane A2 activates platelets through its receptor (TP), a seven‐transmembrane receptor coupled to a heterotrimeric G‐protein, Gq Citation8.
Three isoforms of COXs are so far identified. COX‐1, expressing ubiquitously and constitutively, is the major COX in platelets Citation9–12 (). COX‐2 is an inducible COX expressing in cells at the site of inflammation. COX‐2 is also expressed constitutively in several tissues such as endothelial cells Citation9–12. COX‐3 is a splicing variant of COX‐1, and sensitive to acetoaminophen with little effect on COX‐1 or COX‐2 Citation11. Aspirin, acetylsalicylic acid, is an irreversible inhibitor of COX‐1 and COX‐3, but not COX‐2 at low concentrations. Therefore, the effect of aspirin on platelet function is due to inhibition of the initial step for thromboxane A2 production.
Prostacyclin is produced mainly by the COX‐2 pathway in endothelial cells and inhibits platelet functions (see below) Citation12. The functions of thromboxane A2 and prostacyclin are yin and yang in the regulation of platelet functions. Moreover, cells in vascular walls such as smooth muscle cells express both receptors. Recently, it has been demonstrated that the balance between thromboxane A2 and prostacyclin also regulates atherogenesis since suppression of the thromboxane A2 pathway inhibited atherosclerosis, whereas suppression of the prostacyclin pathway enhanced it in animal models Citation12. Therefore, aspirin would potentially suppress atherogenesis by decreasing the thromboxane A2/prostacyclin ratio.
Aspirin is an antiplatelet drug most widely used in the world. A meta‐analysis by the Antithrombotic Trialist's Collaboration revealed that 22% of cardiovascular events including myocardial infarction, stroke, and vascular death were prevented by aspirin therapy in high‐risk patients Citation13. Aspirin therapy has, thus, become the standard for the secondary prevention of cardiovascular events in high‐risk patients, while it is controversial for the primary prevention in low risk patients.
Aspirin resistance
The efficacy of aspirin in the inhibition of platelet functions differs from patient to patient. Cardiovascular events preferentially occur in patients with low responses to aspirin therapy Citation14–16. This low response is referred to as ‘aspirin resistance’ Citation17–20. Aspirin therapy would bring more effects on the risk reduction if we could find out such patients and modify the therapy by dose‐up or changing aspirin into another drug. However, even the definition of aspirin resistance has not been determined, and the prevalence varies between 5%–60% from report to report depending on laboratory tests used and definitions Citation17.
With the word ‘aspirin resistance’, we should consider biochemical and clinical aspirin resistance Citation10. Biochemical aspirin resistance implies insufficient inhibition of platelet function by aspirin in laboratory tests, while clinical aspirin resistance indicates an increased risk of cardiovascular events in patients under aspirin treatment. Biochemical values in laboratory tests would acquire a strong clinical impact when their correlation with clinical outcome is established. Developing valid and comprehensive values indicating aspirin's efficacy, which predict a cardiovascular risk with a clear cut‐off point, is currently expected.
The light transmission (optical) aggregometer has been conventionally used for assessing aggregability of platelets in platelet‐rich plasma (PRP) Citation21. This method is the standard most widely used in this field. Gum et al. examined platelet aggregability by this method in 326 patients with stable cardiovascular diseases under 325 mg/day aspirin treatment and prospectively followed them for approximately 2 years Citation22. They observed aspirin resistance in 17 cases (5.2%) defined as a mean aggregation of ⩾70% with 10 µM ADP and ⩾20% with 0.5 mg/mL arachidonic acid and a 4.14‐times increase of cardiovascular risk in the aspirin resistant group Citation22.
On the other hand, the optical aggregometer requires preparation of PRP. This procedure may affect the aggregation. Another limitation of this ex vivo examination is evaluating aggregation of platelets alone. In other words, this method eliminates the effects of other blood cells that may affect thrombus formation in vivo. It has been shown that the presence of erythrocytes promotes platelet aggregation Citation23, and leukocytes produce platelet activating factors Citation24.
For a more physiological and simpler procedure, several systems to examine whole‐blood aggregation have been developed. Ultra Rapid Platelet Function assay (URPF)‐ASA (Accumetrics Inc., San Diego, California, USA) is a whole‐blood aggregometer, where whole blood is added to a cartridge containing fibrinogen‐coated beads, and the optical transmission is measured after removing the beads. Using this method, Chen et al. reported that percutaneous coronary intervention (PCI)‐related increase of cardiac troponin I and MB isoenzyme of creatine kinase were more frequently elevated in aspirin resistant patients Citation25.
Thromboxane A2 is an unstable molecule and its stable metabolite, 11‐dehydro‐thromboxane B2, is measured as a value reflecting thromboxane A2 production Citation26. Therefore, it is expected that 11‐dehydro‐thromboxane B2 level is reduced by aspirin intake, and that this value indicates the efficacy of aspirin although thromboxane A2 is produced by other types of cells through the COX‐2 pathway Citation12,Citation17. The Heart Outcomes Prevention Evaluation (HOPE) Study is a randomized, placebo‐controlled, 2×2 factorial trial of ramipril and vitamin E for the secondary prevention of cardiovascular diseases. Reevaluation of the HOPE Study patients treated with aspirin revealed that the highest quartile group of urine 11‐dehydro‐thromboxane B2 level (>33.8 ng/urine mmol creatinine) had a 1.8‐times higher risk for myocardial infarction, stroke and cardiovascular death, compared with the lowest quartile (<15.1 ng/urine mmol creatinine) Citation26. On the other hand, urine 11‐dehydro‐thromboxane B2 levels were widely distributed from individual to individual although they were reduced by aspirin intake in each individual. Urine 11‐dehydro‐thromboxane B2 levels in healthy volunteers were reported to be 72.4–625.9 (179.5±142.0) ng/urine mmol creatinine before aspirin intake, and 12.9–118.0 (39.8±22.9) ng/urine mmol creatinine after aspirin intake Citation27. Further prospective studies would be required to establish the urine 11‐dehydro‐thromboxane B2 level as a marker of aspirin efficacy linking to clinical outcome.
Mechanisms of aspirin resistance
Several mechanisms are considered as causes of aspirin resistance. The dose of aspirin might be insufficient in some patients with aspirin resistance. Platelet Function Analyzer (PFA)‐100 is a whole‐blood aggregometer (Dade Behring, Deerfield, Illinois, USA) that measures the ‘closure time’, a period before closing the special cartridges coated with platelet agonists (collagen plus either ADP or norepinephrine) by clot under a high‐shear conditions Citation28. Using this system, Gonzalez‐Conejero et al. reported that the ‘closure times’ in 29% of 24 subjects were not efficiently prolonged by a 100 mg/day aspirin intake, but completely inhibited by a 500 mg/day aspirin intake Citation27.
Drug interaction could be a cause of aspirin resistance. Non‐steroidal anti‐inflammatory drugs (NSAIDs) are commonly used for their analgesic, anti‐inflammatory, and antipyretic effects. They inhibit not only COX‐2 but also COX‐1. In a cross‐over study in healthy subjects, concomitant administration of ibuprofen was reported to antagonize platelet inhibition induced by aspirin, while that of rofecoxib (a COX‐2 inhibitor), acetoaminophen, or dicrofenac showed no effect Citation29. Since aspirin and NSAIDs bind the same site of COX‐1, the interaction of aspirin with COX‐1 would be competitively inhibited in the presence of other NSAIDs. Aspirin binds COX‐1 irreversibly while ibuprofen does reversibly. Since the lifespan of aspirin is very short, the COX‐1 activity would be recovered after the effect of ibuprofen is lost. This hypothesis is supported by the observation that ibuprofen antagonized aspirin's effect when ibuprofen was administered 2 hours before aspirin, but not 2 hours after aspirin Citation29. The reason why dicrofenac, another NSAID, did not affect aspirin's function is unclear Citation29.
The effect of NSAIDs on the aspirin therapy was examined in 7,107 patients with cardiovascular diseases Citation30. This study revealed that the hazard ratio of the risk of cardiovascular mortality was 1.73 (95% CI: 1.05–2.84) in the aspirin plus ibuprofen group compared to that in the aspirin alone group Citation30. Such increase of risk was not observed in the aspirin plus dicrofenac or aspirin plus other NSAID group Citation30.
Physicians' Health Study, a completed randomized trial of aspirin for the primary prevention of cardiovascular diseases, was reevaluated from the aspect of NSAID intake Citation31. In the aspirin‐treated group, the use of NSAIDs on more than 60 days/year was associated with the risk of myocardial infarction (relative risk (RR) 2.86; 95% CI: 1.25–6.56) compared with no use of NSAIDs. These data suggest that NSAIDs, especially ibuprofen, may reduce the benefit of aspirin for the prevention of cardiovascular diseases.
It has been reported that a 75‐mg daily aspirin intake causes platelet inhibition without attenuation during 2 years, measured by the optical aggregometer using 1 µg/mL collagen as a stimulus Citation32. However, a recent study showed an opposite finding that aspirin's effects progressively decreased during aspirin therapy for a long period Citation33. In this study, the maximal aggregation rates induced by 2 µg/mL collagen at 0, 2 and 24 months after aspirin intake were 88.2±21.8%, 37.9±24.4% and 61.9±23.9%, respectively, while the inhibition of 2 µM ADP‐induced platelet aggregation by ticlopidine did not decrease at 24 months Citation33. Time‐dependent decrease of aspirin's effects might be a cause of aspirin resistance. This mechanism is unknown, but it might be due to compensation by enhanced other signaling pathways in platelets. If this is the case, this phenomenon is clinically quite critical and we would have to modify aspirin therapy after 2‐year treatment. Further study is urgently required.
It has been reported that aspirin did not inhibit platelet aggregation or thromboxane A2 production in vitro at Day 5 or Day 10 after coronary bypass surgery Citation24. COX‐2, a poor substrate of aspirin, was transiently induced in platelets at Day 5. However, expression of COX‐2 was attenuated to the baseline level at Day 10 when the aspirin resistance was still detected. Furthermore, in vitro addition of a selective COX‐2 inhibitor celecoxib did not affect arachidonic acid‐induced thromboxane A2 formation or aggregation of PRP in aspirin‐resistant patients after bypass surgery Citation24. These results suggest that induced COX‐2 may contribute to the aspirin resistance, but it would not be the only factor responsible for the resistance immediately after bypass surgery.
Thromboxane A2 might be produced by a ‘transcellular’ mechanism Citation26. Macrophages and vascular endothelial cells produce prostaglandin H2 from arachidonic acid by COX‐2. This prostaglandin H2 is released to outside of the cells and absorbed into platelets. Then, platelets could utilize prostaglandin H2 as a source of thromboxane A2 production. This type of production of thromboxane A2 would bypass COX‐1.
Furthermore, other habitual and circumstantial conditions also affect the aspirin efficacy. Collagen‐induced platelet aggregability measured under aspirin treatment is positively correlated with serum total cholesterol levels Citation34. Norepinephrine‐induced platelet activation is only partly inhibited by aspirin Citation35. Cigarette smoking increased platelet aggregability in habitual smokers with coronary heart disease and this enhanced aggregability is not inhibited by aspirin Citation36. These reports suggest that high serum cholesterol, physical and mental stress and/or cigarette smoking may cause aspirin resistance in vivo, although their molecular mechanisms remain unclear.
There are more than 100 single nucleotide polymorphisms (SNP) in the thromboxane A2 production pathway, which might be involved in aspirin resistance Citation37,38. SNPs in COX‐1, GPIa, GPIIIa, and FXIII have been shown to affect aspirin's effects on platelet functions Citation27,Citation37,38, while their impact on the clinical outcome remains to be determined.
ADP receptor antagonists
ADP is stored in dense granules and secreted from activated platelets by regulated exocytosis (). Platelets express three types of P2 purinergic receptors: metabotropic ADP receptors P2Y1 and P2Y12, and an ionotropic P2X1 adenosine triphosphate (ATP) receptor involved in calcium ion influx Citation18,Citation39,40. The P2Y1 and P2Y12 receptors are required for ADP‐induced full aggregation, while it remains unclear whether the P2X1 receptor plays a role in platelet aggregation Citation18,Citation39,40. The P2Y1 receptor is associated with a heterotrimeric G‐protein, Gq, which induces phospholipase C activation to cause protein kinase C activation, an increase of intracellular calcium ion concentrations and transient aggregation Citation41. On the other hand, the P2Y12 receptor is coupled to Gi which causes a decrease of the cyclic adenosine monophosphate (cAMP) level and activation of phosphoinositide 3‐kinase Citation40. The P2Y12 signaling is crucial for stabilizing thrombus under high‐shear condition demonstrated by in vitro flow models Citation42,43. Furthermore, in vivo observation of experimental thrombus formation by a real‐time intravital microscopy revealed that blockade of the P2Y12 receptor reduced the initial size of thrombus and frequency of embolization Citation44.
Irreversible P2Y12 antagonists, ticlopidine and clopidogrel, are prodrugs that are activated through oxidation by liver enzyme cytochrome P450 Citation10,Citation18. In several trials, ticlopidine has been demonstrated to reduce cardiovascular events Citation10, Citation45. In the CAPRIE (Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events) Study, patients with vascular diseases were followed under treatment with either clopidogrel (75 mg/day) or aspirin (325 mg/day) for 1–3 years (mean 1.9 years) Citation46. Cardiovascular events occurred in 5.3% in the clopidogrel group and in 5.8% in the aspirin group (P = 0.043) Citation46. This study revealed superior effects of clopidogrel to those of aspirin although the difference was not large. The CURE (Clopidogrel in Unstable angina to prevent Recurrent Event trial) Study demonstrated that treatment with aspirin plus clopidogrel reduced cardiovascular events by 20% compared with aspirin alone Citation47. Thus, P2Y12 blockers have similar, or even stronger, preventive effects for ischemic cardiovascular events compared to aspirin. During treatment with these drugs, especially with ticlopidine, attention should be paid to adverse effects such as liver dysfunction, neutropenia and thrombocytopenic purpura caused by autoimmunity against a vWF‐cleaving enzyme, ADAMTS13 Citation48.
Clopidogrel resistance has also been reported. Sixty patients with acute myocardial infarction were treated with clopidogrel and prospectively followed for 6 months Citation49. Patients were stratified into four groups according to relative reduction of the ADP‐induced maximal aggregation rates at Day 6 compared with those at Day 0 measured by the optical aggregometer. Vascular events occurred in 6 patients out of 15 (40%) in the group with the lowest response to clopidogrel, while in only 1 out of 45 (2.2%) in the other group Citation49. Thus, vascular events preferentially occurred in the clopidogrel‐resistant group.
Furthermore, clopidogrel is a prodrug activated by cytochrome P450 including CYP3A4 in the liver. The inhibitory effects of clopidogrel on platelet aggregation are correlated with activity of CYP3A4 Citation50. Therefore, effects of clopidogrel are inhibited by erythromycin and atorvastatin that are also metabolized by CYP3A4, and enhanced by CYP3A4 inducers such as rifampicin Citation50. Polymorphisms of CYP3A4 and the P2Y12 ADP‐receptor could affect the effect of clopidogrel to inhibit platelet aggregation and might be involved in the clopidogrel resistance.
Drugs that increase intracellular cAMP and cyclic guanosine monophosphate (cGMP)
Prostacyclin, mainly produced by endothelial cells, inhibits platelet aggregation and secretion by elevating intracellular cAMP levels through its specific Gs‐coupling receptor, IP Citation51. Prostacyclin also has a vasodilatory effect. It reduces an ischemic pain in peripheral artery disease (PAD) patients and is used for the treatment of pulmonary hypertension Citation52.
Since the half‐life of prostacyclin is only 3 minutes Citation53, an orally‐mediated prostacyclin‐analogue, beraprost, has been developed as a stable compound whose half‐life is approximately 1 hour Citation54. It inhibits platelet aggregation for several hours after oral intake Citation54. A placebo‐controlled randomized trial enrolling patients with PAD revealed that beraprost improved PAD‐related symptoms such as pain‐free walking distance Citation55, although this improvement was not observed in another study Citation56. However, a meta‐analysis of these trials has shown that beraprost significantly reduced combined endpoints including cardiovascular events and critical events in lower‐limb, such as deterioration of the ischemia and amputation, by 39% Citation57. Other prostacyclin analogues, iloprost and remodulin, have been developed as injection drugs.
Nitric oxide stimulates guanylate cyclase and increases cyclic guanosine monophosphate (cGMP) levels in platelets, causing inhibition of platelet functions Citation58. Effects of cAMP and cGMP are mainly mediated by cAMP‐dependent protein kinase (PKA) and cGMP‐dependent protein kinase (PKG), respectively. Both kinases phosphorylate many proteins in platelets and one of the common PKA and PKG substrates involved in the regulation of platelet aggregation is vasodilator‐stimulated phosphoprotein (VASP), a regulator of actin‐cytoskeletal reorganization Citation59. Recently, the measurement of phosphorylation levels of VASP is under development to monitor the efficacy of antiplatelet drugs affecting PKA and/or PKG Citation59.
Levels of cAMP and cGMP are also regulated by their degrading enzymes cyclic‐nucleotide phosphodiesterases (PDE) Citation58. Major PDEs in platelets are PDE2, PDE3A and PDE5. While PDE5 is specific for cGMP, PDE2 and PDE3A degrade both cAMP and cGMP. There is a feedback regulation among some PDEs. For example, cGMP stimulates the PDE2 activity and inhibits the PDE3A activity Citation58. Importantly, these PDEs are also expressed in cardiovascular systems such as vascular smooth muscle cells and cardiomyocytes. For this reason, PDE‐inhibitors have effects on these cells in addition to platelets.
Cilostazol is an inhibitor of PDE3A, a major cAMP‐degrading enzyme expressed in platelets Citation60,61. Since cilostazol improves symptoms and walking distance in PAD patients Citation62,63, it is recommended for the treatment of PAD Citation64. Recently, cilostazol has been demonstrated to inhibit recurrent ischemic cerebrovascular events by 42% in a prospective randomized trial performed in Japan Citation65. It should be noted, however, that cilostazol increases cAMP levels in cardiomyocytes, causing side effects such as tachycardia in some patients.
Dipyridamole is an inhibitor of PDE5 to increase cGMP levels in platelets and inhibits their aggregation Citation66. European Stroke Prevention Study 2 (ESPS‐2) is a randomized placebo‐controlled double‐blind trial enrolling 6,602 patients with prior cerebral infarction, analyzing the effect of dipyridamole on the secondary prevention of ischemic stroke Citation67. It has demonstrated that dipyridamole alone reduced the stroke risk compared to placebo by 16%, aspirin alone by 18%, and dipyridamole plus aspirin by 36% Citation67, indicating that the efficient risk reduction was obtained by dipyridamole. However, a Cochrane review analyzing 19,842 patients in 26 studies including ESPS‐2 revealed that dipyridamole did not prevent vascular death (RR: 1.02; 95% CI: 0.90–1.17) and that it prevented vascular events (RR: 0.90; 95% CI: 0.83–0.98) although it only reached statistical significance due to the ESPS‐2 results Citation68. Further trials would be required for establishing a preventive effect of dipyridamole.
Omega‐3 fatty acids
It is well known that fish oil contains omega‐3 fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). The GISSI‐Prevention Study has demonstrated that treatment with omega‐3 fatty acids reduced cardiovascular events (death, non‐fatal myocardial infarction and non‐fatal stroke) by 15% in patients with prior coronary heart disease Citation69. The Japan EPA Lipid Intervention Study (JELIS) was performed as a prospective open‐labeled randomized trial where approximately 18,000 hypercholesterolemic patients treated with HMG‐CoA reductase inhibitors were followed with or without EPA (1,800 mg/day) for 5 years Citation70. This study revealed that EPA reduced major coronary events by 19% Citation70, indicating that EPA exerted additional preventive effects for patients treated with cholesterol‐lowering therapy.
Omega‐3 fatty acids not only decrease triglyceride levels but also inhibit platelet functions Citation71. EPA is incorporated into phospholipids, components of cellular membrane, by replacing arachidonic acid. Non‐functional thromboxane A3 is generated from EPA in platelets and functional prostacyclin (PGI3) is produced in endothelial cells Citation72. Therefore, EPA decreases the functional thromboxane A2/prostacyclin ratio, resulting in an inhibition of platelet functions. This effect might contribute to the risk reduction by omega‐3 fatty acids.
Serotonin receptor antagonists
Serotonin (5‐HT) is synthesized in neuronal cells in brain and enterochromaffin cells in the gastrointestinal tract Citation73. Platelets take in enteric cell‐released serotonin through serotonin transporters and store it in dense granules. Serotonin is released from activated platelets at the site of bleeding and vascular damage such as plaque rupture, while neuronal serotonin is stored in specific types of neurons and used as a neurotransmitter.
Serotonin receptors are divided into 7 distinct classes (5‐HT1 to 5‐HT7) and 14 subfamilies. Serotonin secreted from activated platelets plays a role as a self‐agonist to amplify platelet activation and as a vasoconstrictor for vascular smooth muscle cells via the Gq‐coupling 5‐HT2A receptor Citation74. As selective 5‐HT2A receptor antagonists, naftidrofuryl Citation75,76 and sarpogrelate Citation77 have been developed. It has been demonstrated in randomized placebo‐controlled studies Citation75,76 that naftidrofuryl improved clinical symptoms such as walking distance in patients with PAD. Therefore, it is recommended for the treatment of PAD Citation64.
GPIIb/IIIa antagonists
Abciximab, a Fab fragment of a monoclonal antibody recognizing activated integrin GPIIb/IIIa, is used as an adjunctive therapy for percutaneous coronary intervention (PCI) Citation78. Non‐antibody type inhibitors against activated GPIIb/IIIa, eptifibatide and tirofiban, have also been developed as injection drugs and their usefulness has been demonstrated in patients undergoing PCI Citation79,80. Oral GPIIb/IIIa inhibitors were examined by several large‐scale clinical trials. However, the meta‐analysis of these trials revealed that these drugs brought no benefit for reducing coronary events, but increased adverse bleeding events Citation81,82. Their narrow therapeutic range of concentration and partial agonist effects of these drugs might contribute to the results Citation81,Citation83.
Conclusions
Antiplatelet therapy has been established to prevent cardiovascular events in high‐risk patients. It is well known, however, that efficacies of antiplatelet drugs vary from patient to patient. Furthermore, the cardiovascular risk is elevated in patients with platelets resistant to these drugs compared to the good responders, even though the definition of the drug resistance is not yet fixed. Platelet function is not usually monitored in ordinary clinical practice. We need to develop a simple and reliable laboratory test for monitoring the effect of antiplatelet therapy, like blood pressure in antihypertensive therapy and serum cholesterol concentration in cholesterol lowering therapy. A diagnostic value of laboratory examination acquires valid clinical significance when it is associated with clinical outcome. Further prospective clinical trials are required to establish such a value in this field. Several drugs with antiplatelet effects are currently available in addition to aspirin and thienopyridine derivatives, and evidence has been accumulating for these drugs. For patients with insufficient response to an antiplatelet drug, we could improve the therapy by increasing the dose or changing the antiplatelet drug into others.
Acknowledgements
I am grateful to Dr Susana Prat, Barcelona, a visiting scientist in our Department; Dr Yasuyuki Fujita, London; Dr Arata Tabuchi, Berlin; Dr Hidenori Arai, Kyoto; and members of my laboratory for critical reading and discussion of this manuscript. I also thank Professor Dr Toru Kita for continuous encouragement of my research projects. This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Research Grants No. 16209031, by Health and Labour Sciences Research Grant for Cardiovascular Research H17‐006 from the Ministry of Health Labour and Welfare, Japan and in part by grants from the Takeda Science Foundation, Japan Cardiovascular Research Foundation, and a grant from The Kato Memorial Trust for Nambyo Research.
References
- Falk E., Shah P. K., Fuster V. Coronary plaque disruption. Circulation 1995; 92: 657–71
- Shah P. K. New insights into the pathogenesis and prevention of acute coronary syndromes. Am J Cardiol 1997; 79: 17–23
- Brass L. F. The molecular basis of platelet activation. Hematology: Basic Principles and Practice., R Hoffman, E. J Benz, Jr, S. J Shattil, B Furie, H. J Cohen, L. E Silberstein, , editors, et al. Elsevier, New York 2005; p. 1889–914, 4th ed
- Plow E. F., Ginsberg M. H. The molecular basis for platelet funcion. Hematology: Basic Principles and Practice., R Hoffman, E. J Benz, Jr, S. J Shattil, B Furie, H. J Cohen, L. E Silberstein, , editors, et al. Elsevier, New York 2005; p. 1881–97, 4th ed
- Fuster V., Moreno P. R., Fayad Z. A., Corti R., Badimon J. J. Atherothrombosis and high‐risk plaque: part I: evolving concepts. J Am Coll Cardiol 2005; 46: 937–54
- Coughlin S. R. Thrombin signalling and protease‐activated receptors. Nature 2000; 407: 258–64
- Knight D. E., Hallam T. J., Scrutton M. C. Agonist selectivity and second messenger concentration in Ca2+‐mediated secretion. Nature 1982; 296: 256–7
- Hirata T., Ushikubi F., Kakizuka A., Okuma M., Narumiya S. Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest 1996; 97: 949–56
- Patrono C. Aspirin as an antiplatelet drug. N Engl J Med 1994; 330: 1287–94
- Bhatt D. L., Topol E. J. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2: 15–28
- Chandrasekharan N. V., Dai H., Roos K. L., Evanson N. K., Tomsik J., Elton T. S., et al. COX‐3, a cyclooxygenase‐1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A 2002; 99: 13926–31
- Dogne J. M., Hanson J., Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol Sci 2005; 26: 639–44
- Antithrombotic Trialists' Colloboration. Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324: 71–86
- Grotemeyer K. H., Scharafinski H. W., Husstedt I. W. Two‐year follow‐up of aspirin responder and aspirin non responder. A pilot‐study including 180 post‐stroke patients. Thromb Res 1993; 71: 397–403
- Mueller M. R., Salat A., Stangl P., Murabito M., Pulaki S., Boehm D., et al. Variable platelet response to low‐dose ASA and the risk of limb deterioration in patients submitted to peripheral arterial angioplasty. Thromb Haemost 1997; 78: 1003–7
- Grundmann K., Jaschonek K., Kleine B., Dichgans J., Topka H. Aspirin non‐responder status in patients with recurrent cerebral ischemic attacks. J Neurol 2003; 250: 63–6
- Eikelboom J. W., Hankey G. J. Aspirin resistance: a new independent predictor of vascular events?. J Am Coll Cardiol 2003; 41: 966–8
- Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol 2004; 24: 1980–7
- Sztriha L. K., Sas K., Vecsei L. Aspirin resistance in stroke: 2004. J Neurol Sci 2005; 229–30: 163–9
- Michelson A. D., Cattaneo M., Eikelboom J. W., Gurbel P., Kottke‐Marchant K., Kunicki T. J., et al. Aspirin resistance: position paper of the Working Group on Aspirin Resistance. J Thromb Haemost 2005; 3: 1309–11
- Born G. V., Haslam R. J., Goldman M. Comparative Effectiveness of Adenosine Analogues as Inhibitors of Blood‐Platelet Aggregation and as Vasodilators in Man. Nature 1965; 205: 678–80
- Gum P. A., Kottke‐Marchant K., Welsh P. A., White J., Topol E. J. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol 2003; 41: 961–5
- Valles J., Santos M. T., Aznar J., Osa A., Lago A., Cosin J., et al. Erythrocyte promotion of platelet reactivity decreases the effectiveness of aspirin as an antithrombotic therapeutic modality: the effect of low‐dose aspirin is less than optimal in patients with vascular disease due to prothrombotic effects of erythrocytes on platelet reactivity. Circulation 1998; 97: 350–5
- Zimmermann N., Wenk A., Kim U., Kienzle P., Weber A. A., Gams E., et al. Functional and biochemical evaluation of platelet aspirin resistance after coronary artery bypass surgery. Circulation 2003; 108: 542–7
- Chen W. H., Lee P. Y., Ng W., Tse H. F., Lau C. P. Aspirin resistance is associated with a high incidence of myonecrosis after non‐urgent percutaneous coronary intervention despite clopidogrel pretreatment. J Am Coll Cardiol 2004; 43: 1122–6
- Eikelboom J. W., Hirsh J., Weitz J. I., Johnston M., Yi Q., Yusuf S. Aspirin‐resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002; 105: 1650–5
- Gonzalez‐Conejero R., Rivera J., Corral J., Acuna C., Guerrero J. A., Vicente V. Biological assessment of aspirin efficacy on healthy individuals: heterogeneous response or aspirin failure?. Stroke 2005; 36: 276–80
- Nicholson N. S., Panzer‐Knodle S. G., Haas N. F., Taite B. B., Szalony J. A., Page J. D., et al. Assessment of platelet function assays. Am Heart J 1998; 135: S170–8
- Catella‐Lawson F., Reilly M. P., Kapoor S. C., Cucchiara A. J., DeMarco S., Tournier B., et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med 2001; 345: 1809–17
- MacDonald T. M., Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet 2003; 361: 573–4
- Kurth T., Glynn R. J., Walker A. M., Chan K. A., Buring J. E., Hennekens C. H., et al. Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal antiinflammatory drugs. Circulation 2003; 108: 1191–5
- Berglund U., Wallentin L. Persistent inhibition of platelet function during long‐term treatment with 75 mg acetylsalicylic acid daily in men with unstable coronary artery disease. Eur Heart J 1991; 12: 428–33
- Pulcinelli F. M., Pignatelli P., Celestini A., Riondino S., Gazzaniga P. P., Violi F. Inhibition of platelet aggregation by aspirin progressively decreases in long‐term treated patients. J Am Coll Cardiol 2004; 43: 979–84
- Friend M., Vucenik I., Miller M. Research pointers: Platelet responsiveness to aspirin in patients with hyperlipidaemia. BMJ 2003; 326: 82–3
- Larsson P. T., Wallen N. H., Hjemdahl P. Norepinephrine‐induced human platelet activation in vivo is only partly counteracted by aspirin. Circulation 1994; 89: 1951–7
- Hung J., Lam J. Y., Lacoste L., Letchacovski G. Cigarette smoking acutely increases platelet thrombus formation in patients with coronary artery disease taking aspirin. Circulation 1995; 92: 2432–6
- Halushka M. K., Halushka P. V. Why are some individuals resistant to the cardioprotective effects of aspirin? Could it be thromboxane A2?. Circulation 2002; 105: 1620–2
- Cambria‐Kiely J. A., Gandhi P. J. Aspirin resistance and genetic polymorphisms. J Thromb Thrombolysis 2002; 14: 51–8
- Kunapuli S. P., Dorsam R. T., Kim S., Quinton T. M. Platelet purinergic receptors. Curr Opin Pharmacol 2003; 3: 175–80
- Dorsam R. T., Kunapuli S. P. Central role of the P2Y12 receptor in platelet activation. J Clin Invest 2004; 113: 340–5
- Jin J., Daniel J. L., Kunapuli S. P. Molecular basis for ADP‐induced platelet activation. II. The P2Y1 receptor mediates ADP‐induced intracellular calcium mobilization and shape change in platelets. J Biol Chem 1998; 273: 2030–4
- Goto S., Tamura N., Eto K., Ikeda Y., Handa S. Functional significance of adenosine 5'‐diphosphate receptor (P2Y(12)) in platelet activation initiated by binding of von Willebrand factor to platelet GP Ibalpha induced by conditions of high shear rate. Circulation 2002; 105: 2531–6
- Remijn J. A., Wu Y. P., Jeninga E. H., IJsseldijk M. J., van Willigen G., de Groot P. G., et al. Role of ADP receptor P2Y(12) in platelet adhesion and thrombus formation in flowing blood. Arterioscler Thromb Vasc Biol 2002; 22: 686–91
- van Gestel M. A., Heemskerk J. W., Slaaf D. W., Heijnen V. V., Reneman R. S., oude Egbrink M. G. In vivo blockade of platelet ADP receptor P2Y12 reduces embolus and thrombus formation but not thrombus stability. Arterioscler Thromb Vasc Biol 2003; 23: 518–23
- Hass W. K., Easton J. D., Adams H. P., Jr., Pryse‐Phillips W., Molony B. A., Anderson S., et al. A randomized trial comparing ticlopidine hydrochloride with aspirin for the prevention of stroke in high‐risk patients. Ticlopidine Aspirin Stroke Study Group. N Engl J Med 1989; 321: 501–7
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996; 348: 1329–39
- Yusuf S., Zhao F., Mehta S. R., Chrolavicius S., Tognoni G., Fox K. K. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST‐segment elevation. N Engl J Med 2001; 345: 494–502
- Moake J. L. Thrombotic thrombocytopenic purpura: the systemic clumping “plague”. Annu Rev Med 2002; 53: 75–88
- Matetzky S., Shenkman B., Guetta V., Shechter M., Bienart R., Goldenberg I., et al. Clopidogrel resistance is associated with increased risk of recurrent atherothrombotic events in patients with acute myocardial infarction. Circulation 2004; 109: 3171–5
- Lau W. C., Gurbel P. A., Watkins P. B., Neer C. J., Hopp A. S., Carville D. G., et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation 2004; 109: 166–71
- Narumiya S., Sugimoto Y., Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev 1999; 79: 1193–226
- Humbert M., Sitbon O., Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–36
- Dusting G. J., Moncada S., Vane J. R. Recirculation of prostacyclin (PGI2) in the dog. Br J Pharmacol 1978; 64: 315–20
- Demolis J. L., Robert A., Mouren M., Funck‐Brentano C., Jaillon P. Pharmacokinetics and platelet antiaggregating effects of beraprost, an oral stable prostacyclin analogue, in healthy volunteers. J Cardiovasc Pharmacol 1993; 22: 711–6
- Lievre M., Morand S., Besse B., Fiessinger J. N., Boissel J. P. Oral Beraprost sodium, a prostaglandin I(2) analogue, for intermittent claudication: a double‐blind, randomized, multicenter controlled trial. Beraprost et Claudication Intermittente (BERCI) Research Group. Circulation 2000; 102: 426–31
- Mohler E. R 3rd., Hiatt W. R., Olin J. W., Wade M., Jeffs R., Hirsch A. T. Treatment of intermittent claudication with beraprost sodium, an orally active prostaglandin I2 analogue: a double‐blinded, randomized, controlled trial. J Am Coll Cardiol 2003; 41: 1679–86
- Origasa H., Ikeda Y., Shimada K., Shigematsu H. Oral Beraprost Sodium as a Prostaglandin I2 Analogue for Vascular Events in patients with Peripheral Arterial Disease: Meta‐Analysis of Two Placebo‐Controlled Randomized Trials. Jpn J Pharmacoepidemiol 2004; 9: 45–51
- Haslam R. J., Dickinson N. T., Jang E. K. Cyclic nucleotides and phosphodiesterases in platelets. Thromb Haemost 1999; 82: 412–23
- Geiger J., Teichmann L., Grossmann R., Aktas B., Steigerwald U., Walter U., et al. Monitoring of clopidogrel action: comparison of methods. Clin Chem 2005; 51: 957–65
- Sudo T., Tachibana K., Toga K., Tochizawa S., Inoue Y., Kimura Y., et al. Potent effects of novel anti‐platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem Pharmacol 2000; 59347–56
- Goto S. Cilostazol: Potential mechanism of action for antithrombotic effects accompanied by a low rate of bleeding. Atheroscler Suppl 2005; Nov 3, [Epub ahead of print]
- Money S. R., Herd J. A., Isaacsohn J. L., Davidson M., Cutler B., Heckman J., et al. Effect of cilostazol on walking distances in patients with intermittent claudication caused by peripheral vascular disease. J Vasc Surg 1998; 27: 267–74
- Dawson D. L., Cutler B. S., Meissner M. H., Strandness D. E., Jr. Cilostazol has beneficial effects in treatment of intermittent claudication: results from a multicenter, randomized, prospective, double‐blind trial. Circulation 1998; 98: 678–86
- Dormandy J. A., Rutherford R. B. Management of peripheral arterial disease (PAD). TASC Working Group. TransAtlantic Inter‐Society Concensus (TASC). J Vasc Surg 2000; 31: S1–S296
- Gotoh F., Tohgi H., Hirai S., Terashi A., Fukuuchi Y., Otomo E., et al. Cirostazol Stroke Prevention Study: A placebo‐controlled double‐blinded trial for secondary prevention of cerebral infarction. Journal of Stroke and Cerebrovascular Diseases 2000; 9: 147–57
- Sakuma I., Akaishi Y., Fukao M., Makita Y., Makita M. A., Kobayashi T., et al. Dipyridamole potentiates the anti‐aggregating effect of endothelium‐derived relaxing factor. Thromb Res Suppl 1990; 12: 87–90
- Diener H. C., Cunha L., Forbes C., Sivenius J., Smets P., Lowenthal A. European Stroke Prevention Study 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143: 1–13
- De Schryver E. L., Algra A., van Gijn J. Cochrane review: dipyridamole for preventing major vascular events in patients with vascular disease. Stroke 2003; 34: 2072–80
- Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Dietary supplementation with n‐3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI‐Prevenzione trial. Lancet 1999; 354: 447–55
- Yokoyama M. Effects of eicosapentaenoic acid (EPA) on major cardiovascular events in hypercholesterolemic patients: the Japan EPA Lipid Intervention Study (JELIS) Circulation. 2005; 112: 3362–3
- Kris‐Etherton P. M., Harris W. S., Appel L. J. Fish consumption, fish oil, omega‐3 fatty acids, and cardiovascular disease. Circulation 2002; 106: 2747–57
- Needleman P., Raz A., Minkes M. S., Ferrendelli J. A., Sprecher H. Triene prostaglandins: prostacyclin and thromboxane biosynthesis and unique biological properties. Proc Natl Acad Sci U S A 1979; 76: 944–8
- Tyce G. M. Origin and metabolism of serotonin. J Cardiovasc Pharmacol 1990; 16((Suppl 3))S1–7
- Vanhoutte P. M. Platelet‐derived serotonin, the endothelium, and cardiovascular disease. J Cardiovasc Pharmacol 1991; 17((Suppl 5))S6–12
- Trubestein G., Bohme H., Heidrich H., Heinrich F., Hirche H., Maass U., et al. Naftidrofuryl in chronic arterial disease. Results of a controlled multicenter study. Angiology 1984; 35: 701–8
- Adhoute G., Bacourt F., Barral M., Cardon J. M., Chevalier J. M., Cuny A., et al. Naftidrofuryl in chronic arterial disease. Results of a six month controlled multicenter study using Naftidrofuryl tablets 200 mg. Angiology 1986; 37: 160–7
- Doggrell S. A. Sarpogrelate: cardiovascular and renal clinical potential. Expert Opin Investig Drugs 2004; 13: 865–74
- Bhatt D. L., Topol E. J. Current role of platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes. JAMA 2000; 284: 1549–58
- O'Shea J. C., Hafley G. E., Greenberg S., Hasselblad V., Lorenz T. J., Kitt M. M., et al. Platelet glycoprotein IIb/IIIa integrin blockade with eptifibatide in coronary stent intervention: the ESPRIT trial: a randomized controlled trial. JAMA 2001; 285: 2468–73
- Topol E. J., Moliterno D. J., Herrmann H. C., Powers E. R., Grines C. L., Cohen D. J., et al. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med 2001; 344: 1888–94
- Quinn M. J., Plow E. F., Topol E. J. Platelet glycoprotein IIb/IIIa inhibitors: recognition of a two‐edged sword?. Circulation 2002; 106: 379–85
- Quinn M. J., Byzova T. V., Qin J., Topol E. J., Plow E. F. Integrin alphaIIbbeta3 and its antagonism. Arterioscler Thromb Vasc Biol 2003; 23: 945–52
- Cox D., Smith R., Quinn M., Theroux P., Crean P., Fitzgerald D. J. Evidence of platelet activation during treatment with a GPIIb/IIIa antagonist in patients presenting with acute coronary syndromes. J Am Coll Cardiol 2000; 36: 1514–9