Abstract
Today, the average life expectancy in developed nations is over 80 years and climbing. And yet, the quality of life during those additional years is often significantly diminished by the effects of age‐related, degenerative diseases, including age‐related macular degeneration (AMD), the leading cause of blindness in the elderly worldwide. AMD is characterized by a progressive loss of central vision attributable to degenerative and neovascular changes in the macula, a highly specialized region of the ocular retina responsible for fine visual acuity. Estimates gathered from the most recent World Health Organization (WHO) global eye disease survey conservatively indicate that 14 million persons are blind or severely visually impaired because of AMD. The disease has a tremendous impact on the physical and mental health of the geriatric population and their families and is becoming a major public health burden.
Currently, there is neither a cure nor a means to prevent AMD. Palliative treatment options for the less prevalent, late‐stage ‘wet’ form of the disease include anti‐neovascular agents, photodynamic therapy and thermal laser. There are no current therapies for the more common ‘dry’ AMD, except for the use of antioxidants that delay progression in 20%–25% of eyes.
New discoveries, however, are beginning to provide a much clearer picture of the relevant cellular events, genetic factors, and biochemical processes associated with early AMD. Recently, compelling evidence has emerged that the innate immune system and, more specifically, uncontrolled regulation of the complement alternative pathway plays a central role in the pathobiology of AMD. The complement Factor H gene—which encodes the major inhibitor of the complement alternative pathway—is the first gene identified in multiple independent studies that confers a significant genetic risk for the development of AMD. The emergence of this new paradigm of AMD pathogenesis should hasten the development of novel diagnostic and therapeutic approaches for this disease that will dramatically improve the quality of our prolonged lifespan.
Clinical principles
Age‐related macular degeneration (AMD) is the most common cause of legal blindness among people over 60 years. Clinical hallmarks of the disease are observed in at least one third of persons over age 75 in industrialized countries.
AMD is the leading cause of blindness in the elderly worldwide, affecting 30–50 million individuals. Conservatively, the World Health Organization estimated in 2002 that 14 million persons worldwide are blind or severely visually impaired because of AMD.
AMD is often grouped into two basic clinical categories: the ‘dry’ atrophic form and the exudative neovascular, or ‘wet’, form.
There is neither a preventive therapy nor a cure for AMD. Palliative treatment options for wet AMD include anti‐neovascular agents, photodynamic therapy and thermal laser. There are no current therapies for dry AMD, although antioxidant therapy delays progression in about 20%–25% of eyes.
Pathophysiologic principles
The complement alternative pathway plays a predominant role in AMD pathology.
Allelic variations in genes coding the complement alternative pathway‐associated proteins Factor H and Factor B confer risk for or protection from AMD. Combined analysis of the haplotypes for the genes encoding these proteins indicates that variation in the two loci can predict the clinical outcome in 74% of AMD cases.
Environmental factors modulate genetic susceptibility for the condition. Smoking and obesity are the two strongest risk factors identified to date.
New discoveries related to the underlying genetic susceptibility for AMD open new avenues for its diagnosis, treatment and prevention.
Introduction
Age‐related macular degeneration (AMD), the leading cause of worldwide blindness in the elderly, is a bilateral ocular condition that affects the central area of retina known as the macula. The macula—which derives its name from the deposition of xanthophyll pigments—is located temporal to the optic disc and is bounded by the temporal superior and inferior vascular arcades ( and ). Although the macula comprises only 4% of retinal area, it is responsible for the majority of useful photopic vision. The fovea lies at the center of the macula and is approximately 2 mm in diameter (, and ). This region contains the highest density of cone photoreceptor cells and is the only region of the retina where 20/20 vision is attainable. Although the macula is only 6 mm in diameter, it accounts for almost 10% of the entire visual field. Thus, lesions developing in this region can have a major impact on visual function.
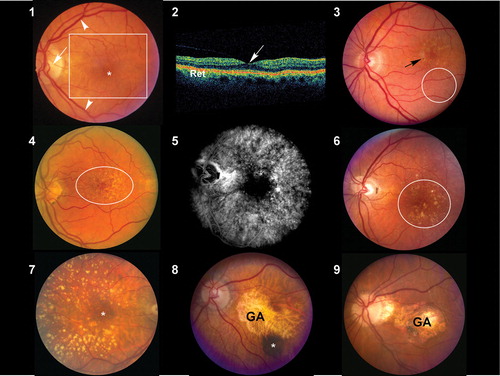
Figures 1–9. : Normal macula of an elderly patient. The asterisk represents the location of the fovea, which lies directly in the visual axis. The macula (boxed area), which is adapted for high acuity vision, is located temporal to the optic nerve (arrow). It is approximately 6 mm in diameter and centered on the fovea. The vascular arcades are indicated by arrowheads. : Ocular coherence tomogram (OCT) of a normal macula. The central area of depression (arrow) represents the fovea, corresponding to the asterisk depicted in . Ret = retina. : Color fundus photograph derived from an individual with early, dry age‐related macular degeneration (AMD). Retinal pigment epithelium (RPE) pigment disruption is present in the macula (arrow) and numerous small (<63 microns in diameter) hard drusen are present inferior to this region (oval). : Color fundus photograph from an individual with dry AMD. Numerous small and intermediate‐sized drusen are visible in the macular region (oval). and : Corresponding fluorescein angiogram () and color fundus photograph () images from an individual with dry AMD. Although larger and confluent drusen are visible in the color image (circle), the number and extent of drusen distribution is appreciated even more clearly in the angiogram. The hyperfluorescent drusen observed in the angiogram are of uniform size and often referred to as cuticular drusen. : Color fundus photograph from an individual with dry AMD, depicting the presence of numerous large (>125 micron diameter), calcified drusen deposited primarily within the peri‐ and parafoveal regions. Smaller drusen are present in the foveal region (asterisk). and : Color fundus photograph from two patients with macular geographic atrophy (GA). The margins of the regions of RPE atrophy are clearly delineated. Choroidal blood vessels are more easily visualized in these regions of atrophy because of the loss and/or absence of the RPE pigment. A choroidal nevus (asterisk) is indicated in . These eyes would be expected to have poor central vision due to the extensive atrophy.
AMD has a tremendous impact on the physical and mental health of members of the geriatric population and their families. Prior to 1990, AMD of all forms was often referred to as ‘senile macular degeneration’ or SMD, a reflection of the fact that the vision loss associated with AMD manifests late in life when most affected individuals are looking forward to enjoying retirement activities and maintaining independence. Instead, millions with AMD suffer bilateral central vision loss such that they can no longer drive, read a newspaper, prepare meals, or enjoy recreational activities. For many patients, the visual impairment associated with AMD means a loss of independence, depression, increased financial concerns and the need to adapt to vision loss at a time when co‐morbid conditions often make such adaptation difficult or impossible Citation1–5.
The cost to society is only now being appreciated. A recent analysis of AMD in Australia predicts that the disease costs $2.6 billion per year Citation6. This is projected to grow to $6.5 billion by 2025, a total cost of $59 billion over the next 20 years. A treatment that reduced the progression by only 10% would save Australia $5.7 billion over that same period of time. Similar analyses for the United States are lacking, but given the demographics and higher cost of medical care in the US, the costs would be projected to as much as 20‐fold higher.
Clinically, AMD is classified into the nonexudative ‘dry’ or atrophic form and the exudative ‘wet’ or neovascular form. More severe vision loss is typically associated with the ‘wet’ form that occurs in about 15% of all patients with AMD, but up to 20% of legal blindness from AMD is due to the atrophic form Citation7.
This article reviews the epidemiological, clinical, and underlying genetic and pathological manifestations of AMD.
Clinical aspects of AMD
Epidemiology
AMD is the leading cause of blindness in the elderly worldwide, affecting 30–50 million individuals. World Health Organization (WHO) statistics from the most recent WHO global eye disease surveys conducted in 2002 revealed that 8.7% of worldwide blindness is due to AMD, the third leading cause of worldwide blindness after cataract and glaucoma. Conservatively, the WHO estimated that 14 million persons worldwide are blind or severely visually impaired because of AMD Citation8,9. The majority of these impaired individuals and the millions more who are visually impaired from this condition live in developed countries.
The Beaver Dam Eye Study, a large population‐based study, revealed that the prevalence of large extracellular ocular deposits known as drusen, a hallmark sign of AMD, is 2% in persons 43–54 years of age and 24% in persons over 75 years of age in the United States Citation10. Data from a number of large epidemiological studies indicate that almost one in three people over the age of 70 have early stages of AMD and that the incidence of the more severe forms of the disease increases from less than 1% in people under 65 years of age to over 25% in the 90 plus age group Citation11–13.
The Age‐related Eye Disease Study (AREDS) found that 20.2% of individuals with early stage AMD progressed to advanced disease over a 5‐year period, a rate of 4.0% per year. Similar annual rates of progression, between 2.5% and 4.8%, were reported in the Rotterdam Study and Blue Mountains Eye Study (BMES) Citation14,15. A recent report by Taylor and colleagues estimates that the progression rate for mild to moderate visual impairment is approximately 32%, and the rate from moderate to severe visual impairment is 46%, based upon the 2–3‐year progression data derived from the Macular Photocoagulation Study (MPS), the Treatment of AMD with Photodynamic Therapy (TAP) study and the Vertiporfin in Photodynamic Therapy (VIP) study Citation6. These rates are slightly higher than the rates of 16% and 17% per annum reported by the MVIP study. Taylor also showed that the incidence of mild visual impairment in individuals who are 40 years and older is 0.10% per year; and, for moderate and severe visual impairment in this same group, it is 0.04% and 0.11% per year, respectively. However, as life expectancies increase in all parts of the world the prevalence of AMD will increase. It is likely that in the next few decades, tens of millions of people worldwide will suffer legal blindness or severe visual impairment from AMD Citation16–24.
Given the enormous impact of AMD on an aging population, much public interest and research has been focused on this condition in the past decade. Recent advances in the treatment of the ‘wet’ or neovascular form of the disease provide new treatment options for those who already suffer from AMD. Moreover, recent groundbreaking discoveries of the genetic allelic variations that predispose individuals to the disease provide the opportunity to develop methods of preclinical diagnosis and preventive therapies for early stage disease.
Clinical presentation of AMD
Although autopsy studies have documented histological and ultrastructural changes associated with AMD in the retina, retinal pigment epithelium, choriocapillaris and choroid in middle age (see below), the disease typically does not manifest clinically before age 55 Citation10. AMD is generally thought to progress along a continuum from atrophic or ‘dry’ AMD to neovascular ‘wet’ AMD with approximately 10%–15% of all AMD patients eventually developing the wet form. Occasionally patients with no prior signs of dry AMD present with exudative changes as the first manifestation of the condition. The pathognomonic clinical sign of ‘dry’ AMD is pigment disruption and drusen—small yellowish deposits observed on fundus examination. Drusen may be small ‘hard’ (small with discrete margins) or ‘soft’ (larger with indistinct edges) (–). They lie between the retinal pigment epithelium (RPE) and an adjacent basement membrane complex known as Bruch's membrane (, –). Geographic atrophy (GA) and RPE changes are also observed in dry AMD (–). Several classification schemes have been developed that subdivide dry AMD into categories based on the number and size of drusen, amount of GA and degree of pigmentary changes in the macula Citation25–28.
The designation of exudative or ‘wet’ AMD implies that fluid, exudates and/or blood are present in the extracellular space between the neural retina and the RPE (i.e. the subretinal space) and/or, as in the case of RPE detachments, between the RPE and Bruch's membrane (i.e. the sub‐RPE space). Angiography using sodium fluorescein dye is commonly performed when signs of AMD are observed and exudative changes are suspected. Angiography can demonstrate ‘classic’ lacey vessels or more ‘occult’ leakage associated with fibrovascular tissue lying in the subretinal or sub‐RPE space (–). The neovascular tissue associated with exudative AMD is most commonly referred to as choroidal neovascularization (CNV) because it originates from the normal choriocapillaris and extends through a dehiscence in Bruch's membrane into the subretinal or sub‐RPE space ( and ). Sometimes multiple soft drusen become confluent and create large pigment epithelial detachments (PEDs), which represent an elevation of the retinal pigment epithelium under the retina ( and ). Neovascularization associated with PEDs can be difficult to visualize clinically or image using fluorescein angiography; therefore, it is difficult to treat Citation29.
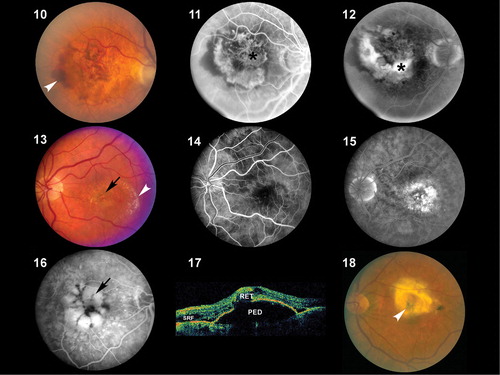
Figures 10–18. Color fundus photograph(), early fluorescein angiogram () and late fluorescein angiogram () from an individual with a classic choroidal neovascularization (CNV). Subretinal blood and fluid (arrowhead depicts edge) is clearly visible within the macular region in . Note the lacey appearance of vessels (asterisk) in the early angiogram. Marked leakage of fluorescein (asterisk), with indistinct edges of hyperfluorescence (white area) is clearly visible in the late angiogram. Color fundus photograph (), early fluorescein angiogram () and late fluorescein angiogram () from an individual with occult choroidal neovascularization. A single, small punctate region of hemorrhage (black arrow) and a ring of exudates (arrowhead depicts edge) that appears more indolent than that which typically occurs in classic choroidal neovascularization (CNV) () is visible in . Note that there is minimal hyperfluorescence in the early phase of the angiogram, in contrast to that observed in the early stage of classic CNV. Speckled macular hyperfluorescence–‐in contrast to the profuse leakage of fluorescein that occurs in classic lesions–‐is visible in the late stage angiogram (). Corresponding angiographic () and ocular coherence tomogram (OCT) images () from an individual with a macular pigment epithelial detachment (PED). A multilobulated, hyperfluorescent lesion with sharply demarcated borders (arrow) is clearly visible in the late stage angiogram depicted in . The blister‐like elevation of the retina and retinal pigment epithelial layer, which correlates with the PED shown in , is appreciated on OCT in . SRF = subretinal fluid; RET = retina. : Color fundus photograph from an individual with end stage (cicatricial) exudative age‐related macular degeneration (AMD). A large disciform scar (arrowhead) covering the macular region is distinctly visible.
The natural history of dry AMD is progressive, with gradual loss of visual function that may span many years' time. Many patients with dry AMD are asymptomatic and unaware of the disease. In 10%–15% percent of patients with dry AMD the deterioration is more rapid and extensive and they suffer significant vision loss due to geographic atrophy. In approximately 10%–15% of patients the condition progresses to the ‘wet’ or neovascular form. Left untreated, the natural history of wet AMD advances further to a cicatricial stage referred to as a disciform scar. This process usually takes place over several months and typically results in a 4–8‐mm diameter fibrotic scar underlying the macula accompanied by a central scotoma with severe central vision loss in one eye ( and ). Patients with neovascular AMD in one eye have a 4%–12% per year cumulative risk of developing neovascular AMD in the fellow eye. Thus, the risk of bilateral loss of central vision is high in those with the neovascular form of the disease Citation30.
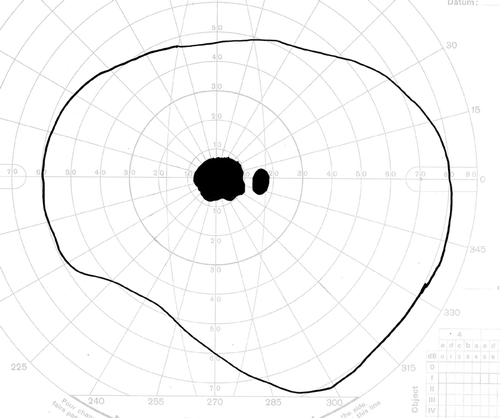
Figure 19. Goldmann visual field simulating an individual with central visual loss. The physiological blind spot, which corresponds to the location of the optic nerve head, is depicted as a small dark oval. The larger, more irregular darkened region corresponds to a blind spot, or scotoma, caused by atrophy and/or scarring within the macula.
Therapeutics
A precise understanding of the molecular pathogenesis of AMD has proven to be elusive but it has only recently begun to be elucidated. As such, it is not surprising that the therapeutic approaches for AMD developed over the past 30 years have been of only limited benefit to most patients. Therapeutic interventions have focused almost exclusively upon the exudative ‘wet’ form that comprises approximately 10% of the AMD patient population because it is the most debilitating and rapidly progressive form of the disease. Until recently, such therapies have resulted in a benefit to less than 50% of patients with ‘wet’ AMD, and therefore less than 5% of all AMD patients. Efforts aimed at developing therapeutics to delay onset or progression of ‘dry’ AMD (or the conversion from ‘dry’ to ‘wet’ AMD) have proven even more difficult due to the long study duration and large numbers of participants required to achieve meaningful statistical results.
Over the last 30 years epidemiological studies have repeatedly identified four risk factors for AMD: age, cigarette smoking, increased body mass index, and inheritance Citation10,Citation31,32. These same studies, as well as others, have produced conflicting information regarding the roles of dietary antioxidant supplementation and fat intake. In 1992 the Age‐related Eye Disease Study (AREDS) group began enrolling subjects in a prospective, multicenter, randomized, controlled study in which 4757 subjects aged 55–80 years with various stages of AMD were randomized to placebo or combinations of antioxidant therapy Citation33. Patients enrolled in the AREDS had either mild, moderate, or severe dry AMD in both eyes, or exudative AMD in one eye only. The study was designed to determine whether antioxidant therapy prevented vision loss, slowed the progression of dry AMD, or prevented conversion from dry to wet AMD. The results of 6.3 years of follow‐up in the AREDS study were published in 2001 Citation33. The study results demonstrated that daily antioxidant therapy in the form of 15 mg beta carotene, 500 mg vitamin C, 50 mg vitamin E, 80 mg zinc (as zinc oxide), and 2 mg copper (as cupric oxide) daily was superior to a placebo in delaying progression of advanced dry AMD, and in slowing conversion from dry to wet AMD. Specifically, the odds ratio of progression to advanced AMD was decreased to 0.72 in the group who took the above combination of antioxidants as compared to placebo. No benefit of antioxidant supplementation was demonstrated in the milder forms of AMD. However, because the early stages of AMD often persist for many years, it is quite possible that the study design was not long enough to demonstrate a benefit to patients with milder stages of AMD. The AREDS was a landmark study in that it was the first and largest prospective randomized trial designed to analyze the effect of antioxidant supplementation on the progression of AMD.
Another therapeutic intervention that has been investigated for nonexudative AMD is prophylactic laser therapy for eyes with drusen. Three prospective trials, the Choroidal Neovascularization Prevention Trial (CNVPT), Complications of AMD Prevention Trial (CAPT) and Prophylactic Treatment of Age‐Related Macular Degeneration Trial (PTAMD), evaluated the efficacy and safety of light macular grid laser in reducing the risk of developing CNV in eyes with drusen. The CNVPT and CAPT used argon laser and the PTAMD used an 810‐nm diode laser Citation34–36. In both the CNVPT and PTAMD studies, grid laser treatment increased the risk of CNV. Therefore grid laser treatment of eyes with drusen is not recommended if the fellow eye has CNV Citation36,37. Recent results of the PTAMD bilateral drusen study arm with 639 subjects reported no decrease in CNV development but a four‐letter gain in visual acuity in treated versus control eyes after 3 years of follow‐up Citation38. The larger CAPT study of treatment of one eye in the setting of bilateral drusen is ongoing but near completion of 5 years of follow‐up. Until the results of the CAPT study are known, prophylactic laser treatment of eyes with bilateral drusen is not a recommended therapy.
Therapy for exudative AMD has evolved from thermal laser photocoagulation in the early to mid 1980s, to photodynamic therapy for CNV beginning in 2000 and, most recently, to inhibition of vascular growth factors. The 3‐year results of the Macular Photocoagulation Studies (MPS) for AMD was published in 1986 and demonstrated a benefit to thermal laser photocoagulation for ‘extrafoveal’ CNV located between 200 and 3,000 microns from the fovea. Subsequent MPS studies for juxtafoveal (1–200 microns from the fovea) CNV and subfoveal CNV, published in 1990 and 1991, respectively, demonstrated a benefit for laser treatment versus no treatment Citation39–41. However, visual loss was common even in those eyes with successful obliteration of CNV Citation42. Additionally, the closer the CNV was to the fovea, the poorer the visual outcome and the higher the rate of recurrence even in those who initially responded to treatment Citation43. In 1993, Freund and co‐workers demonstrated that only 13% of new patients with exudative AMD (or approximately 1.3% of all AMD patients) met criteria for thermal laser treatment based on the MPS Citation44.
In the late 1980s to mid 1990s there was significant interest in vitreoretinal surgical approaches to exudative AMD. Variable success rates were reported in numerous small pilot trials of vitrectomy surgery with evacuation of subretinal blood and neovascular membranes. Trial designs were nonstandard, results were conflicting, and long‐term follow‐up in these pilot trials was lacking. Given the potential morbidity and expense of a major eye surgery, the National Eye Institute sponsored the prospective, randomized, controlled Submacular Surgery Trials (SST), two arms of which evaluated the safety and efficacy of removal of submacular CNV and blood in exudative AMD. The results of the SST for AMD demonstrated that surgery (as performed in the trial) did not increase the chance of stable or improved vision, and it prevented severe vision loss only in those eyes with large subretinal hemorrhages. Submacular surgery was also associated with high risk of rhegmatogenous retinal detachment. Given these results, submacular surgery is no longer recommended for exudative AMD Citation45,46.
Other surgical therapeutic modalities which have been investigated in small pilot trials are macular translocation, reported first by Machemer and Steinhorst, and limited macular translocation developed later by de Juan Citation47,48. The former technique involves creation of a total retinal detachment, while the latter is limited to a partial retinal detachment. Both techniques involve displacement of the macular retina from an area of CNV to a location where the retinal pigment epithelium is anticipated to be healthier. While pilot trials have suggested a benefit from these therapies for a small subset of patients, the morbidity of the major eye surgery involved in an elderly population, the lack of results from definitive prospective, randomized controlled trials, and the emergence of less invasive and more broadly applicable pharmacological therapies for exudative AMD have resulted in much less frequent application of macular translocation in recent years.
In 2000, photodynamic therapy with VisudyneTM (verteporfin), a light‐activated compound, was approved by the Food and Drug Administration (FDA) for exudative AMD with predominately (>50%) classic, subfoveal CNV that was no larger than 5600 microns in diameter. FDA approval was based on the results of the Treatment of Age‐Related Macular Degeneration with Photodynamic Therapy Investigation (TAP) study. In the TAP study, subfoveal CNV was divided into subgroups based on initial visual acuity, lesion size, and fluorescein leakage characteristics: enrolled subjects were randomized to VisudyneTM or placebo Citation49. Photodynamic therapy involves intravenous administration of the photoactivatable compound, VisudyneTM, followed 15 minutes later by irradiation of the CNV with a low‐energy, nonthermal laser. Activated VisudyneTM generates singlet oxygen that damages the CNV endothelium resulting in thrombosis Citation50. In the TAP study an average of 3.5 treatments spaced at 3‐month intervals were required to ablate the CNV, so the process is a lengthy one. Visual results, though better than with any prior treatment, were less than optimal with only 67% of treated patients with predominately classic CNV losing fewer than three lines of visual acuity (as compared to 39% of placebo treated eyes). At 1 year, 16% of Visudyne‐treated patients gained one or more line of vision, and 15% of treated patients lost more than six lines of visual acuity. Over the course of the next several years, subgroup analysis of data from the TAP studies as well as additional trials with VisudyneTM treatment for lesions other than predominately classic CNV led to its application for other lesion types. However, the visual results for lesions that were predominantly occult CNV were less favorable than those with predominately classic CNV Citation51,52.
Other ablative treatments for CNV include transpupillary thermotherapy (TTT) and radiation therapy. TTT utilizes low energy diode laser (810 nm) applied to the CNV slowly over 1 minute. Small pilot trials of TTT for occult CNV initially showed promise but a randomized, prospective clinical trial (TTT4 CNV) demonstrated no significant benefit. Similarly, low‐dose radiation therapy for AMD was investigated in multiple small pilot trials with conflicting results Citation53–55. Although no definitive prospective clinical trial of radiation therapy for AMD has ever been conducted, the concept has become less popular in recent years, especially in light of the interest in pharmacological therapies for AMD.
The current era of therapy for exudative AMD utilizes pharmacologic administration of anti‐neovascular agents periocularly or intraocularly. Studies implicating various cellular growth factors, including vascular endothelial growth factor (VEGF) in a variety of ocular neovascular processes, have led to the development of VEGF inhibitors, VEGF antibodies and other ‘broad‐spectrum’ antiangiogenic molecules that inhibit proangiogenic cytokines. In 2004, MacugenTM (pegaptanib sodium) was the first of the VEGF inhibitors to be approved by the FDA for exudative AMD. Pegaptanib is an aptamer which binds to the A isoform of VEGF‐165 at its heparin binding site and prevents VEGF‐165 from binding to its receptor on endothelial cells. Pegaptanib is administered by intravitreal injection (i.e. injected into the vitreous cavity of the eye) every 6 weeks for up to a 2‐year course of therapy. Based on the MacugenTM study results of 1186 patients after 1 year of subjects treated with 0.3 mg pegaptanib, 70% lost fewer than three lines of vision compared to 55% of controls. The risk of severe vision loss (>6 lines of vision) was reduced from 22% in controls to 10% in treated subjects Citation56.
Another VEGF inhibitor undergoing clinical trials is LucentisTM. LucentisTM is derived from the Fab fragment of an antibody to isoform A of VEGF. The fragment blocks VEGF binding to its receptor on endothelial cells, thus inhibiting its biological activity. The 2‐year results of the ANCHOR trial were presented recently Citation57. The data demonstrated not only a preservation of vision in nearly 95% of patients treated with LucentisTM (ranibizumab) at year one but, for the first time, an improvement in vision in a significant proportion of patients with exudative AMD. LucentisTM was FDA‐approved for the treatment of exudative AMD in June 2006.
AvastinTM (bevacizumab), a VEGF antibody derived from the same parent antibody as LucentisTM, is approved by the FDA to treat colon cancer and is gaining popularity as an ‘off‐label’ intravitreal therapy for AMD. It is important to note that AvastinTM has not yet undergone the toxicity studies and controlled trials that are required by the FDA for ocular application Citation58,59. Other antiangiogenic therapies currently under study include anecortave acetate (RetaaneTM)—an antiangiogenic steroid Citation60,61—squalene, triamcinolone (which carries a significant risk of causing cataract and steroid‐induced glaucoma) in combination with VisudyneTM photodynamic therapy Citation62,63, and various VEGF inhibitors that are in various stages of development.
One concern with all antiangiogenic agents is their potential for systemic inhibition that may produce cardiovascular or cerebrovascular complications, or inhibit wound healing. The immediate and long‐term systemic effects of these drugs, especially those prescribed ‘off‐label’, are largely unknown and difficult to determine in an elderly population already prone to vascular events. Additional drawbacks to the intravitreally administered therapeutics include patient discomfort, intraocular infection (endophthalmitis), retinal tear or detachment, and lens damage in phakic patients Citation64.
Recent discoveries of the role that inflammation plays in AMD pathogenesis (see below) has led to an interest in investigating the anti‐inflammatory effects of 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG CoA reductase) inhibitors (statins) on AMD. Several clinical population‐based studies of the effects of statins on AMD development and progression have yielded conflicting results Citation65. Thus, more studies in this area will be required to assess efficacy.
In the near future, therapy for exudative AMD will likely involve multiple anti‐neovascular agents, possibly coupled with less frequent application of ablative therapies such as photodynamic therapy, in order to maximize the beneficial effects of each and minimize the frequency of adverse side effects. Further on the horizon are antiangiogenic agents in longer‐acting forms that require less frequent administration. The ultimate therapy for AMD, however, will lie in preclinical identification of those who are genetically ‘at risk’ for the disease, coupled with preventative strategies that are designed to minimize or compensate for one's genetic predisposition.
Pathobiology of early AMD
Overview
Until quite recently, the majority of research on AMD and the development of therapeutics for the disease have focused upon late‐stage neovascular events. No clear‐cut sequence for the initiation and progression of AMD has been identified that would allow it to be monitored and treated in its earliest stages. Moreover, the pathologic correlates of the different clinical phenotypes of AMD remain poorly understood, and there is a paucity of animal models that accurately mimic the characteristic features of the disease. Nevertheless, recent discoveries are beginning to provide a much clearer picture of the relevant cellular events, genetic factors, and biochemical processes that are associated with early AMD. This new information should hasten significantly the development of clinically effective diagnostics and therapeutics for the treatment of this devastating condition.
Morphological correlates of early AMD
From a histopathologic standpoint, the earliest detectable changes associated with AMD occur at the interface between the macular retina and the underlying layer of connective tissue known as the choroid Citation66–70. At this location lie the outer segments of rod and cone photoreceptors, the retinal pigment epithelial (RPE) cells, a stratified basement membrane complex termed Bruch's membrane (BM), and the choroidal capillary bed or choriocapillaris ().
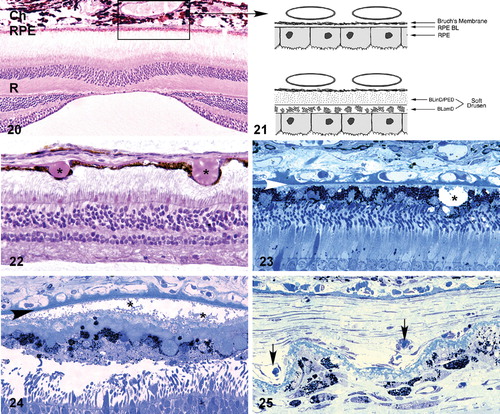
Figures 20–25. Light microscopic(, –) and schematic () images depicting the choroid‐RPE‐retina interface. A section showing the normal anatomical relationships of the macular choroid (Ch), retinal pigmented epithelium (RPE) and neural retina (R) is shown in ; the section passes directly through the foveal pit. The boxed region corresponds to that depicted in , which compares the choriocapillaris‐RPE interface in unaffected (top) and affected (bottom) individuals. The majority of early age‐related macular degeneration (AMD)‐associated extracellular lesions–‐including drusen, basal laminar deposit (BLamD), and basal linear deposits (BLinD)–‐form along this interface. Drusen (asterisks), which form between the RPE and Bruch's membrane, are present in and . Extensive accumulations of BLamD (asterisks), which form between the RPE and its basal lamina, are depicted in . Choroidal neovessels, located in both the sub‐RPE and subretinal spaces (arrows) are shown in . Arrowheads in and , Bruch's membrane.
The initial clinical diagnosis of early AMD is based on the funduscopic identification of drusen—the hallmark indicators of disease—and/or pigmentary changes in the macula. Drusen present funduscopically as yellow‐white deposits (–). They are extracellular deposits located between the retinal pigment epithelial (RPE) basal lamina and the inner collagenous layer of the elastin‐containing Bruch's membrane ( and ) Citation71. Macular drusen are classified using criteria that include size, shape and topographical location. These deposits are associated with an increased risk of visual loss that leads to end stage or late disease through either the growth of new blood vessels (choroidal neovascular membranes; and ) that destroy the macula, or through the development of frank retinal atrophy Citation72–75.
Figures 26–29. Transmission electron micrographs of the retinal pigment epithelium(RPE)‐choroid interface in eyes of donors without () and with (–) a clinically documented history of age‐related macular degeneration (AMD). A single druse (D) is shown in ; its location between the RPE basal lamina (arrowheads) and Bruch's membrane (BM) is clearly indicated. BLamD (asterisk) accumulates between the basal surface of the RPE and its basal lamina (arrow), whereas BLinD is located within the innermost aspect of Bruch's membrane (). A patent choroidal neovessel (asterisk), lying between BM and a layer of BLamD (rectangle), is shown in . RPE = retinal pigment epithelium; BM = Bruch's membrane.
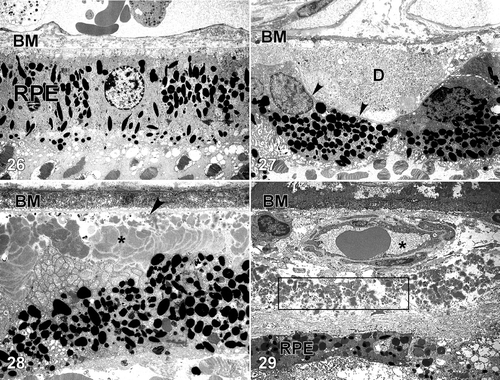
Bruch's membrane consists of an elastin core flanked on both sides by a collagenous layer and a basal lamina (–) Citation76–79. It is widely believed that the barrier properties of the RPE and BM limit cellular migration, especially the invasion of neovascular tissue from the choroid into the subretinal space. Our understanding of the molecular composition of BM, the molecular traffic that occurs across it, and the homeostatic mechanisms that maintain it, during normal aging as well as in AMD, is still rudimentary Citation80.
Links between AMD and structural abnormalities in BM have been documented in numerous histopathologic studies. Classically, these BM defects have been described as fragmentation or fracturing in association with calcification Citation81–85. Additional age‐related changes in BM are typified by: 1) progressive thickening of the two collagenous layers; 2) modification and degeneration of collagen and elastin; 3) increased levels of advanced glycation end products, noncollagenous proteins and lipids; and 4) accumulation of several types of sub‐RPE deposits Citation86–95. It has also been suggested that the age‐related abnormalities in BM eventually lead to photoreceptor degeneration as a result of increased hydrophobicity, reduced permeability, and impaired nutrient exchange between the choroid and the RPE. Functionally, such changes result in an exponential reduction in the hydraulic conductivity of BM as a function of age Citation96–99. Much less is known about changes in BM hydraulic conductivity in individuals with AMD.
It has been proposed that topographic variations in BM may render the macula more susceptible to the ingrowth of new blood vessels from the choroidal vasculature that characterizes neovascular AMD. Morphometric data indicate that the elastic lamina of Bruch's membrane in the macula is 3–6‐fold thinner and 2–5‐fold less dense relative to that in the mid‐periphery in individuals of all ages Citation100. Elastin fiber destruction in the macula could also play a key role in the initiation of neovascular events because elastin degradation peptides are highly angiogenic and possess macrophage‐recruiting activity Citation101,102.
In addition to drusen, basal laminar deposits (BLamD) and basal linear deposits (BLinD) accumulate within the extracellular environment of the RPE‐choroid interface (, , and ). These are identified based upon their morphological appearance and location. Both types of deposits have been proposed by various investigators to be involved in the pathogenesis of AMD Citation103. Neutral lipid (including esterified cholesterol) and apolipoproteins B and E are abundant in these basal deposits and in drusen, in the eyes of normal aged donors, as well as those with a history of AMD Citation88,Citation94,Citation104–107.
Basal laminar deposits (BLamD) that accumulate between the RPE basal plasma membrane and its basal lamina (, and ) do not appear to be specific to or ‘diagnostic’ for AMD; however, there is a strong relationship between macular BLamD and exudative AMD, disciform scarring, and visual loss Citation74,Citation84,Citation108–114. BLamD‐like deposits are also seen in other macular disorders with a clinical appearance similar to what is observed in AMD. These include Sorsby's fundus dystrophy, late onset retinal degeneration (LORD), adult foveomacular pigment epithelial dystrophy and Malattia leventinese Citation115. The molecular composition and origin of BLamD have not yet been identified Citation116–118, but their morphological similarity to long‐spacing collagen is striking. Electron microscopy has revealed that their banding pattern matches that of type VI collagen Citation119.
BLinD lie between the RPE basal lamina and the elastic layer of Bruch's membrane (). The principal component of BLinD is a distinct form of membranous debris, thought to be comprised primarily of lipoprotein particles containing neutral lipids, including esterified cholesterol. It is likely that BLinD are derived from local ocular sources (e.g. RPE cells) Citation120. BLinD were initially described by Sarks and colleagues Citation121 as lipid‐like vesicles with varying diameters.
It is believed that the pigmentary changes often observed in the macula of AMD eyes are attributable to degenerative changes in the highly melanized RPE cells. This notion has gained wide acceptance because most of the early clinical signs and histopathological changes have been localized to this cell layer Citation122–125. It has long been recognized that the RPE is essential for the transport of ions, nutrients, and metabolites from the circulation to the neural retina and vice versa Citation126. A viable RPE is also essential for normal photoreceptor cell metabolism and functioning of the visual cycle. Therefore, it is not surprising that RPE degeneration is accompanied by concomitant photoreceptor degeneration. RPE dysfunction in the macula, whether caused by local environmental insults and/or genetic defect(s), would have a profound impact on the overlying photoreceptors and, therefore, on central vision. However, the nature of the insult(s) that causes RPE dysfunction has yet to be ascertained, although a variety of hypotheses has been advanced over the years including genetic factors, ischemia, oxidative stress, phagocytic overload, cigarette smoke, lipofuscin toxicity and, most recently, microbial infection Citation127–133.
The choriocapillaris has also been suspected to play a role in the etiology of AMD. The density and diameters of the choriocapillaris capillaries decrease with age, and this decrease is even greater in patients with AMD. The overall loss appears most marked in regions of geographic atrophy Citation134,135.
Physiological correlates of early AMD
There is substantial evidence of photoreceptor involvement in early AMD, particularly in those patients with soft or large drusen in the macula Citation136–144. Contrast sensitivity, the rate of recovery after photostress, the amplitude and latency of the foveal electroretinogram (ERG) response, and dark adaptation are all affected adversely in early AMD patients, including those with normal visual acuity Citation139,Citation145–150. It has been shown that photoreceptor cells in areas impacted by drusen exhibit morphologic and biochemical signs of degeneration, including decreased expression of synapse‐associated proteins and increased expression of stress‐response molecules. There are also drusen‐associated reductions in photoreceptor cell densities, thus suggesting that degenerative changes in photoreceptors ultimately lead to cell death Citation151,152.
Inflammation and AMD
Recent studies of the molecular composition of drusen have implicated local inflammation as a key element in the pathogenesis of AMD Citation153–159. Drusen contain numerous proteins related to the process of inflammation or its aftermath Citation106,Citation160–163. In particular, many of these proteins are associated with the complement cascade and its regulation. Some of the proteinaceous components in drusen and the sub‐RPE space are activated complement components and fragments associated with assembly of the membrane attack complex (MAC) Citation161,Citation164–167. Other drusen components including vitronectin Citation160, clusterin, complement receptor 1 (CR1), and membrane cofactor protein (MCP‐1) Citation163 are known complement regulatory proteins. Still others include known activators of the complement cascade including cholesterol Citation168, C‐reactive protein Citation154, and the amyloidogenic peptide amyloid β Citation152,Citation169. Metallic zinc, an amyloid β binding molecule, is also a drusen constituent Citation170. This compositional profile forms the basis for the conclusion that drusen are a manifestation of chronic, local inflammation at the level of Bruch's membrane.
The complement system and AMD
Historically the immune system has been partitioned into the ‘innate’ and ‘adaptive’ systems. The innate system, an evolutionarily ancient system present in some form in all multicellular organisms, is designed to respond immediately to invading pathogens. In contrast, the adaptive system, which is specific to vertebrates, takes several days to mobilize T and B lymphocytes in order to contain an infection. As one arm of the innate system, the complement system of proteins is poised to recognize, attack and kill invading microorganisms Citation171–176. The complement cascade consists of the classical, alternative, and lectin activation pathways, which converge on a final common, or terminal, pathway. The complement system is part of a complex homeostatic mechanism that includes recognition and killing of microorganisms, removal of foreign particles and dead cells, recruitment and activation of inflammatory cells, regulation of antibody production and elimination of immune complexes. In addition to the 30 or more proteins that contribute to the complement cascade, there are numerous soluble and membrane‐bound factors that help to modulate the system and play important roles in protecting host cells and tissues from complement attack.
The complement cascade is activated by an initiating event or ‘trigger’. The ‘trigger’ initiates a series of biochemical reactions that produce biologically active peptides and other molecules that generate inflammatory, immune, and physiological responses. The terminal pathway culminates in the assembly of a macromolecular complex known as the membrane attack complex (MAC). The MAC promotes cell lysis by the insertion of transmembrane pores, and the formation of additional byproducts through enzymatic cleavage. These byproducts have inflammatory, anaphylotoxic, and chemotactic properties, which can produce damage to both foreign pathogens and host cells.
Each of the three complement pathways can be distinguished by the nature of the ‘trigger’ that activates it. Typically, antigen‐antibody binding triggers the ‘classical pathway’. In the ‘lectin pathway’, surface carbohydrate residues of invading pathogens serve as binding sites for the proinflammatory, acute phase proteins that trigger the cascade. A low level of spontaneous activation known as ‘tickover’ characterizes the ‘alternative pathway’, the most rudimentary and nonspecific pathway. This continuous background level of activation is amplified rapidly by the recognition of ubiquitous components present on the surfaces of many bacteria, viruses, as well as constituents present in cigarette smoke.
The complement system—and particularly the alternative pathway—plays a key role in tissue injury in ischemia, infection, and in a variety of disease processes Citation177. Under normal conditions, local complement activation is beneficial by promoting the rapid clearance of cell debris and facilitating the removal of toxic protein aggregates Citation178. Uncontrolled activation of this system, however, can seriously damage host cells and tissues and contribute significantly to disease progression Citation178–181. Activated components of the alternative pathway, for example, have been described in the pathology of glomerulonephritis, IgA nephropathy, asthma, lupus, rheumatoid arthritis, myositis, inflammatory central nervous system diseases like multiple sclerosis, cardiac valvular disease, cirrhosis, adult respiratory distress syndrome (ARDS), pemphigoid disease, and antiphospholipid syndrome Citation182–188. Thus, because of its spontaneous activating properties, continuous control of the alternative pathway is necessary to prevent damage to self tissues. This control can be severely affected by local changes in the concentrations of available components or impaired function of regulatory proteins that are attributable to acquired or genetic deficiencies.
Complement Factor H, the complement alternative pathway and AMD
Factor H is the main soluble inhibitor of the alternative pathway and, like most complement components, cells in the liver are responsible for 80%–90% of Factor H protein synthesis Citation189–191. Factor H inhibits C3 activation by binding to C3b, leading to its proteolytic cleavage and decay of the C3 convertase complex (C3bBb). It also competes with Factor B for surface‐bound C3b, thereby conferring protection from membrane attack complex (MAC)‐mediated attack. We now know that variants in the gene encoding Factor H play a central role in one's susceptibility to AMD (see below). We also know from previous studies that individuals with mutations in the Factor H gene, or with autoantibodies that block Factor H function, are particularly susceptible to kidney disease that often leads to renal failure Citation181. However, it is not yet apparent how predicted functional alterations in the protein isoform encoded by the AMD risk‐conferring Factor H gene variant lead to damage in the macula; nor is it known how the corresponding Factor B isoform produces the same net effect. Nevertheless, all of the available data are consistent with the conclusion that uncontrolled activation of the alternative pathway of complement at the level of Bruch's membrane is a key element in the process of drusen formation and a major contributing factor to the pathogenesis of AMD.
Although the liver is responsible for most circulating Factor H, the eye is also capable of producing it as well as some other complement components. Not surprisingly, Factor H protein is also a molecular constituent of drusen Citation192. It co‐localizes with its ligand C3b (a complement pathway component) in substructural spherules within drusen that contain amyloid β, further implicating these structures as candidate complement activators Citation169,Citation193. Factor H and the MAC co‐distribute in drusen and at the interface between the RPE and choroid. Finally, Factor H and C5b‐9 (MAC) immunolabeling are more intense in the macula compared to tissues from more peripheral areas in the same eye (Hageman and Mullins, unpublished).
We have advanced a working model of AMD pathobiology based upon our studies Citation154,Citation194 and the work of others Citation156,Citation195. In this model, RPE atrophy and the subsequent deposition of cellular debris in the sub‐RPE space are construed as a local pro‐inflammatory ‘seeding’ event, leading to activation of the innate immune system at the RPE‐choroid interface. Complement attack, in turn, induces significant bystander damage to macular cells and tissues, thus rendering them susceptible to additional RPE atrophy, photoreceptor degeneration and CNV.
Molecular genetics of AMD and disease associations
Overview
AMD is often regarded as a group of complex, late onset diseases modulated by the convergence of multiple risk factors Citation196,197. Family history is a consistent risk factor identified in most epidemiological studies of AMD conducted to date, and familial aggregation studies have shown that a genetic contribution is readily identified in up to 25% of AMD cases Citation198. Twin studies also support a genetic basis for the disease, with the concordance of clinical features (drusen and pigmentary changes) for both early and late onset disease being approximately twice as high in monozygotic (identical) twins compared to dizygotic (nonidentical) twins Citation199,200.
Linkage analysis and candidate gene screening have recently provided dramatic new insights into the molecular genetics of AMD Citation201–209. Genome‐wide linkage analyses of extended families with AMD have identified a number of chromosomal loci that are linked to AMD. The most consistent of these occurs at chromosome 1q31 Citation210. Between 15% and 40% of multiplex AMD families segregated with a disease gene in the 1q31 region Citation204. Further analysis of this region revealed an allelic variant in exon 104 of the fibulin‐6 gene (FBLN‐6) in members of one AMD family, as well as in some sporadic AMD cases Citation211.
A number of other candidate genes have also been linked to AMD. Several of these associated with a small, but increased risk of developing AMD. However, most of these results have not yet been replicated in subsequent studies. These genes include ELOVL4 Citation212, VEGF, VLDLR, and LRP6Citation213, FIBLN5Citation214 and TLR‐4 Citation215.
In contrast, the ϵ4 allele of the apolipoprotein E (APOE) gene has consistently been shown to be protective for AMD Citation216–218, whereas the ϵ2 allele appears to influence progression and to cause an earlier age of onset Citation218. Reliable association of a gene that confers increased risk for AMD, ABCA4, has also been reported and substantiated Citation219.
In early 2005, in a striking example of how disparate methods for gene discovery can be brought to bear on a single problem, four groups reported independently that common variants (single nucleotide polymorphisms (SNPs)) in the gene encoding complement Factor H (CFH) confer major susceptibility to, or protection from, AMD Citation192,Citation220–222. Individuals who possess a single copy of the risk‐conferring CFH haplotype have 2–4‐fold higher lifetime risk of developing AMD, and those with two copies possess a 5–7‐fold higher lifetime risk. Haplotype analysis showed that a SNP in the CFH gene that results in a tyrosine to histidine substitution at amino acid 402 in the Factor H protein can account for up to 50% of AMD cases in the human population, a number approaching 50,000,000 worldwide. Follow‐up reports have since confirmed these findings in additional cohorts in the United States Citation212,Citation223, Iceland Citation224, the United Kingdom Citation225, and France Citation226. Interestingly, the major CFH risk haplotype in the Japanese population appears to be different than that reported in other populations, although the protective haplotype is the same Citation227.
In early 2006, allelic variants in two other complement‐related genes, Factor B (BF) and complement component C2, were linked to AMD Citation228. These are paralogous genes located 500 bp apart on chromosome 6p21, and both reside in the major histocompatibility complex (MHC) class III region. BF is a component of the alternative pathway of complement, whereas C2 is a component of the classical pathway. Analysis of AMD‐associated SNPs in these two genes identified a single ‘risk’ and two ‘protective’ haplotypes. Functional evidence suggests that the protective SNP possesses a glutamine (Q) in place of the more common arginine (R) at position 32 of the BF protein. There is evidence that this substitution may result in reduced alternative pathway‐mediated hemolytic activity Citation229 and, therefore, a reduced overall propensity for overactivation of complement.
Combined analyses of CFH and BF haplotypes showed that the two variants together can account for nearly 74% of the risk of developing AMD. The protective haplotype for each of the three genes was completely absent in nearly 75% of all AMD cases, whereas 56% of controls possessed at least one copy of a protective CFH or BF haplotype. Approximately 60% of the risk in AMD cases, and 65% of the protection in controls, can be assigned to the CFH gene locus alone.
In addition to the 1q31 locus, a region on chromosome 10q26 has been identified in several recent studies that consistently showed linkage to AMD Citation210 on a scale similar to that of the CFH Tyr402His variant. PLEKHA1 (pleckstrin homology domain‐containing, family A, member 1) and a predicted LOC387715 gene were identified as the relevant candidate genes at that locus Citation230. A subsequent study concluded that SNP rs10490924 in LOC387715 was the most likely AMD susceptibility allele Citation231. A third study identified a coding change in the LOC387715 gene as the significant allele that results in a nonsynonymous substitution of alanine to serine at position 69 of the hypothetical protein. The variant was found to be statistically independent of the increased risk conferred by the CFH allele, and of similar magnitude. In addition, a significant statistical relationship between the LOC387715 variant and a history of cigarette smoking has been found Citation232. Thus far, there is no published evidence for ocular expression of the LOC387715 gene product at either the RNA or protein levels, nor is there any indication of its likely functional properties.
Association of AMD with other diseases
The predicted complement‐regulatory dysfunction associated with AMD is consistent with the pattern of complement‐regulatory dysfunction in other diseases. Under normal conditions, local complement activation promotes the rapid clearance of cellular debris and facilitates removal of toxic protein aggregates. However, uncontrolled activation results in collateral damage to surrounding host tissues. Organ damage from alternative complement dysregulation occurs in several renal diseases including atypical hemolytic uremic syndrome (aHUS) Citation233–239 and membranoproliferative glomerulonephritis type II (MPGNII) Citation240,241. In aHUS, widespread blood clot formation and reactive endothelial cell proliferation in small blood vessels can lead to acute renal failure. Approximately 50% of patients with aHUS have a mutation in one of three complement regulators: Factor H, membrane cofactor protein, or Factor I Citation239. In aHUS, missense mutations in CFH give rise to a protein with altered binding to C3b, thus restricting Factor H in its capacity to protect host cells from complement attack. In MPGNII, uncontrolled systemic activation of the alternative complement pathway leads to the deposition of activated C3 complement fragments within the glomerular basement membrane. Remarkably, many patients with MPGNII also develop early onset macular drusen that are clinically, histologically, ultrastructurally and histochemically indistinguishable from ocular drusen in AMD Citation153. It was this observation that led our group to explore the role of complement dysregulation in AMD, and that ultimately led to the discovery of the CFH gene polymorphisms Citation192.
Although the discovery of a link between AMD and polymorphisms in the CFH and BF genes is a major advance in our understanding of the genetic basis of AMD, major unresolved issues remain. Possession of the BF and/or CFH risk‐conferring haplotypes appears highly likely to place RPE and choroidal cells in the macula at sustained risk for alternative pathway‐mediated complement attack. In the aging eye, it can be envisaged that this eventually leads to drusen accumulation, disruption of Bruch's membrane integrity, and ensuing complications such as geographic atrophy and choroidal neovascularization. However, the nature of the activation event(s) that triggers the complement system in the macula has yet to be ascertained. Based upon the analogy to aHUS and MPGNII, microbial infection of the RPE/choroid is a distinct possibility Citation242, but it is certainly not the only one. It is puzzling why the macula of the eye and the glomerulus in the kidney appear to be the tissues that are most susceptible. Is it because the macula and the glomerulus share anatomical features and/or functional properties that render them more vulnerable to the effects of complement dysregulation? It is also puzzling why the macula is more vulnerable relative to more peripheral retinal locations. Clearly, the biological basis for the macula's predilection for degeneration and neovascularization will require further study. In that regard, it is noteworthy that macular involvement is associated with a number of other ocular and nonocular disorders including macular edema, presumed ocular histoplasmosis syndrome, Alport's syndrome, rubella, ocular toxoplasmosis, bacterial endocarditis, pseudoxanthoma elasticum and Menke's disease Citation29,Citation243.
Conclusion
Complement Factor H is the first gene identified in multiple independent studies that confers a significant risk for the development of AMD. This finding, together with the subsequent identification of AMD‐associated variants in the related complement genes BF and C2, provides compelling evidence that the innate immune system and, more specifically, uncontrolled regulation of the alternative pathway of complement, plays a central role in the pathobiology of AMD. At this point, the most likely scenario is that exposure to infection or some other triggering event in genetically susceptible individuals, coupled with impaired complement regulatory function, leads to the sustained activation of the complement cascade, drusen formation and, eventually, development of AMD.
The emergence of this new paradigm of AMD pathogenesis sets the stage for the rapid development of early diagnostics, novel bioassays, and new animal models that faithfully mimic aspects of the disease process. Furthermore, the way is now paved for development of novel therapeutic interventions aimed at modulating the alternative pathway of complement in ‘at‐risk’ individuals prior to the onset of choroidal neovascularization or geographic atrophy.
Now that the genetic basis for a major proportion of AMD cases has been elucidated, what additional scientific progress may be anticipated in the next few years, and what will be the ramifications of these new findings for the diagnosis and treatment of AMD? In the near term, a more comprehensive understanding of the genetic basis of AMD should rapidly emerge. It will not be surprising, for example, if genetic polymorphisms in additional complement components, complement regulators and, possibly, immune system effectors and inflammatory mediators are implicated in AMD. Secondly, it may be anticipated that the gene variants linked to AMD also contribute to other prevalent age‐related diseases where chronic, local inflammatory processes are involved. For example, a significant statistical relationship between the CFH Tyr402His ‘risk’ variant and the incidence of myocardial infarction has recently been reported Citation242,Citation244. Based upon this new genetic information, it will now be possible to devise genetic screening tests that will identify those individuals who are most at risk of developing AMD later in life. This will enable clinicians to monitor susceptible individuals from an early age, and to develop and test new preventive treatments in the early stages of the disease before the onset of vision loss. Finally, the development of new diagnostic and pharmacological approaches will be hastened by the identification of the alternative pathway of complement as a prime therapeutic target. As our understanding of the pathogenesis of AMD continues to improve rapidly, so do the prospects for novel diagnostic and therapeutic approaches that will dramatically improve the quality of our prolonged lifespan.
Acknowledgements
Supported in part by NIH EY11515 (GH), EY11527 (LJ) and EY11521 (DA), the American Macular Degeneration Foundation (GH), the International Retina Research Foundation (GH), Eye Research Institute (GH), and an unrestricted grants to the University of Iowa from Research to Prevent Blindness, Inc. GH currently holds honorary professorships in the School of Medicine, Queen's University, Belfast and the Shandong Eye Institute, Qingdao, China.
References
- Uhlmann R. F., Larson E. B., Koepsell T. D., Rees T. S., Duckert L. G. Visual impairment and cognitive dysfunction in Alzheimer's disease. J Gen Intern Med 1991; 6: 126–32
- Lin M. Y., Gutierrez P. R., Stone K. L., Yaffe K., Ensrud K. E., Fink H. A., et al. Vision impairment and combined vision and hearing impairment predict cognitive and functional decline in older women. J Am Geriatr Soc 2004; 52: 1996–2002
- Rovner B. W., Casten R. J., Tasman W. S. Effect of depression on vision function in age‐related macular degeneration. Arch Ophthalmol 2002; 120: 1041–4
- Abyad A. In‐office screening for age‐related hearing and vision loss. Geriatrics 1997; 52: 45–6; 51–4; 57
- Berman K., Brodaty H. Psychosocial effects of age‐related macular degeneration. Int Psychogeriatr 2006; 8: 1–14
- Taylor H., Guymer R., Keeffe J. The Impact of Age‐Related Macular Degeneration. University of Melbourne, Melbourne 2006; 1–72, Access Economics Pty Limited
- Sunness J. S. The natural history of geographic atrophy, the advanced atrophic form of age‐related macular degeneration. Mol Vis 1999; 5: 25
- Magnitude and causes of visual impairment. World Health Organization, Fact Sheet No 282.
- Resnikoff S., Pascolini D., Etya'ale D., Kocur I., Pararajasegaram R., Pokharel G. P., et al. Global data on visual impairment in the year 2002. Bull World Health Organ 2004; 82: 844–51
- Klein R., Peto T., Bird A., Vannewkirk M. R. The epidemiology of age‐related macular degeneration. Am J Ophthalmol 2004; 137: 486–95
- Smith W., Assink J., Klein R., Mitchell P., Klaver C., Klein B., et al. Risk factors for age‐related macular degeneration: Pooled findings from three continents. Ophthalmology 2001; 108: 697–704
- Mitchell P., Smith W., Attebo K., Wang J. Prevalence of age‐related maculopathy in Australia. The Blue Mountains Eye Study. Ophthalmology 1995; 102: 1450–60
- VanNewkirk M., Nanjan M., Wang J., Mitchell P., Taylor H., McCarty C. The prevalence of age‐related maculopathy: the visual impairment project. Ophthalmology 2000; 107: 1593–600
- Wolfs R. C., Borger P. H., Ramrattan R. S., Klaver C. C., Hulsman C. A., Hofman A., et al. Changing views on open‐angle glaucoma: definitions and prevalences—The Rotterdam Study. Invest Ophthalmol Vis Sci 2000; 41: 3309–21
- Mitchell P., Wang J. J., Foran S., Smith W. Five‐year incidence of age‐related maculopathy lesions: the Blue Mountains Eye Study. Ophthalmology 2002; 109: 1092–7
- Friedman D. S., O'Colmain B. J., Munoz B., Tomany S. C., McCarty C., de Jong P. T., et al. Prevalence of age‐related macular degeneration in the United States. Arch Ophthalmol 2004; 122: 564–72
- Taylor H. R., Keeffe J. E., Vu H. T., Wang J. J., Rochtchina E., Pezzullo M. L., et al. Vision loss in Australia. Med J Aust 2005; 182: 565–8
- Jonasson F., Arnarsson A., Peto T., Sasaki H., Sasaki K., Bird A. C. 5‐year incidence of age‐related maculopathy in the Reykjavik Eye Study. Ophthalmology 2005; 112: 132–8
- Andersen N. Age‐related macular degeneration among the Inuit in Greenland. Int J Circumpolar Health 2004; 63(Suppl 2)320–3
- Vingerling J. R., Dielemans I., Hofman A., Grobbee D. E., Hijmering M., Kramer C. F., et al. The prevalence of age‐related maculopathy in the Rotterdam Study. Ophthalmology 1995; 102: 205–10
- Krishnaiah S., Das T., Nirmalan P. K., Nutheti R., Shamanna B. R., Rao G. N., et al. Risk factors for age‐related macular degeneration: findings from the Andhra Pradesh eye disease study in South India. Invest Ophthalmol Vis Sci 2005; 46: 4442–9
- Munoz B., Klein R., Rodriguez J., Snyder R., West S. K. Prevalence of age‐related macular degeneration in a population‐based sample of Hispanic people in Arizona: Proyecto VER. Arch Ophthalmol 2005; 123: 1575–80
- Leske M. C., Wu S. Y., Hennis A., Nemesure B., Yang L., Hyman L., et al. Nine‐year incidence of age‐related macular degeneration in the Barbados Eye Studies. Ophthalmology 2006; 113: 29–35
- Wong T. Y., Loon S. C., Saw S. M. The epidemiology of age related eye diseases in Asia. Br J Ophthalmol 2006; 90: 506–11
- Klein R., Davis M. D., Magli Y. L., Segal P., Klein B. E., Hubbard L. The Wisconsin age‐related maculopathy grading system. Ophthalmology 1991; 98: 1128–34
- Age‐Related Eye Disease Study Research Group. The Age‐Related Eye Disease Study (AREDS): design implications. AREDS report no. 1., Control Clin Trials. 20. 573–600
- Davis M. D., Gangnon R. E., Lee L. Y., Hubbard L. D., Klein B. E., Klein R., et al. The Age‐Related Eye Disease Study severity scale for age‐related macular degeneration: AREDS Report No.17. Arch Ophthalmol 2005; 123: 1484–98
- Ferris F. L., Davis M. D., Clemons T. E., Lee L. Y., Chew E. Y., Lindblad A. S., et al. A simplified severity scale for age‐related macular degeneration: AREDS Report No.18. Arch Ophthalmol 2005; 123: 1570–4
- Wray S., Kuwabara T., Sanderson P. Menkes' kinky hair disease: a light and electron microscopic study of the eye. Invest Ophthalmol 1976; 15: 128–38
- Abugreen S., Muldrew K. A., Stevenson M. R., VanLeeuwen R., DeJong P. T., Chakravarthy U. CNV subtype in first eyes predicts severity of ARM in fellow eyes. Br J Ophthalmol 2003; 87: 307–11
- Clemons T. E., Milton R. C., Klein R., Seddon J. M., Ferris F. L., 3rd. Risk factors for the incidence of Advanced Age‐Related Macular Degeneration in the Age‐Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology 2005; 112: 533–9
- Thornton J., Edwards R., Mitchell P., Harrison R. A., Buchan I., Kelly S. P. Smoking and age‐related macular degeneration: a review of association. Eye 2005; 19: 935–44
- Age‐Related Eye Disease Study Research Group. A randomized, placebo‐controlled, clinical trial of high‐dose supplementation with vitamins C and E, beta carotene, and zinc for age‐related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001; 119: 1417–36
- Choroidal neovascularization in the Choroidal Neovascularization Prevention Trial, The Choroidal Neovascularization Prevention Trial Research Group. Ophthalmology 1998; 105: 1364–72
- Complications of Age‐Related Macular Degeneration Prevention Trial Study Group. The Complications of Age‐Related Macular Degeneration Prevention Trial (CAPT): rationale, design and methodology. Clin Trials 2004; 1: 91–107
- Friberg T. R., Musch D. C., Lim J. I., Morse L., Freeman W., Sinclair S. Prophylactic treatment of age‐related macular degeneration report number 1: 810‐nanometer laser to eyes with drusen. Unilaterally eligible patients. Ophthalmology 2006; 113: 622 e1
- Prenner J. L., Rosenblatt B. J., Tolentino M. J., Ying G. S., Javornik N. B., Maguire M. G., et al. Risk factors for choroidal neovascularization and vision loss in the fellow eye study of CNVPT. Retina 2003; 23: 307–14
- Friberg T. R., the PTAMD Study Group. The Prophylactic Treatment of AMD Multi‐Centered Trial (PTAMD): Results From the Bilateral Study Arm. Invest Ophthalmol Vis Sci 2006; 47, E‐Abstract 3538
- Argon laser photocoagulation for neovascular maculopathy, Three‐year results from randomized clinical trials. Macular Photocoagulation Study Group. Arch Ophthalmol 1986; 104: 694–701
- Krypton laser photocoagulation for neovascular lesions of age‐related macular degeneration, Results of a randomized clinical trial. Macular Photocoagulation Study Group. Arch Ophthalmol 1990; 108: 816–24
- Laser photocoagulation of subfoveal neovascular lesions in age‐related macular degeneration, Results of a randomized clinical trial. Macular Photocoagulation Study Group. Arch Ophthalmol 1991; 109: 1220–31
- Han D. P., Folk J. C., Bratton A. R. Visual loss after successful photocoagulation of choroidal neovascularization. Ophthalmology 1988; 95: 1380–4
- Persistent and recurrent neovascularization after krypton laser photocoagulation for neovascular lesions of age‐related macular degeneration, Macular Photocoagulation Study Group. Arch Ophthalmol 1990; 108: 825–31
- Freund K. B., Yannuzzi L. A., Sorenson J. A. Age‐related macular degeneration and choroidal neovascularization. Am J Ophthalmol 1993; 115: 786–91
- Bressler N. M., Bressler S. B., Childs A. L., Haller J. A., Hawkins B. S., Lewis H., et al. Surgery for hemorrhagic choroidal neovascular lesions of age‐related macular degeneration: ophthalmic findings: SST report no. 13. Ophthalmology 2004; 111: 1993–2006
- Hawkins B. S., Bressler N. M., Miskala P. H., Bressler S. B., Holekamp N. M., Marsh M. J., et al. Surgery for subfoveal choroidal neovascularization in age‐related macular degeneration: ophthalmic findings: SST report no. 11. Ophthalmology 2004; 111: 1967–80
- Machemer R., Steinhorst U. H. Retinal separation, retinotomy, and macular relocation: II. A surgical approach for age‐related macular degeneration? Graefes Arch Clin Exp Ophthalmol 1993; 231: 635–41
- de Juan E., Jr., Loewenstein A., Bressler N. M., Alexander J. Translocation of the retina for management of subfoveal choroidal neovascularization II: a preliminary report in humans. Am J Ophthalmol 1998; 125: 635–46
- Photodynamic therapy of subfoveal choroidal neovascularization in age‐related macular degeneration with verteporfin: one‐year results of 2 randomized clinical trials—TAP report. Treatment of age‐related macular degeneration with photodynamic therapy (TAP) Study Group. Arch Ophthalmol 1999; 117: 1329–45
- Schmidt‐Erfurth U., Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age‐related macular degeneration. Surv Ophthalmol 2000; 45: 195–214
- Verteporfin In Photodynamic Therapy Study Group. Verteporfin therapy of subfoveal choroidal neovascularization in age‐related macular degeneration: two‐year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization—verteporfin in photodynamic therapy report 2. Am J Ophthalmol 2001; 131: 541–60
- Bressler N. M., Arnold J., Benchaboune M., Blumenkranz M. S., Fish G. E., Gragoudas E. S., et al. Verteporfin therapy of subfoveal choroidal neovascularization in patients with age‐related macular degeneration: additional information regarding baseline lesion composition's impact on vision outcomes—TAP report No.3. Arch Ophthalmol 2002; 120: 1443–54
- Stevenson M. R., Hart P. M., Chakravarthy U., Mackenzie G., Bird A. C., Owens S. L., et al. Visual functioning and quality of life in the SubFoveal Radiotherapy Study (SFRADS): SFRADS report 2. Br J Ophthalmol 2005; 89: 1045–51
- Goverdhan S. V., Gibbs F. A., Lotery A. J. Radiotherapy for age‐related macular degeneration: no more pilot studies please. Eye 2005; 19: 1137–41
- Marcus D. M., Peskin E., Maguire M., Weissgold D., Alexander J., Fine S., et al. The age‐related macular degeneration radiotherapy trial (AMDRT): one year results from a pilot study. Am J Ophthalmol 2004; 138: 818–28
- Gragoudas E. S., Adamis A. P., Cunningham E. T., Jr., Feinsod M., Guyer D. R. Pegaptanib for neovascular age‐related macular degeneration. N Engl J Med 2004 30; 351: 2805–16
- Heier JS., Shapiro H., Singh AA. Randomized, Controlled Phase III Study of Ranibizumab (LucentisTM) for Minimally Classic or Occult Neovascular Age‐Related Macular Degeneration: Two‐Year Efficacy Results of the MARINA Study. Invest Ophthalmol Vis Sci 2006; 47, E‐Abstract 2959
- Rosenfeld P. J., Moshfeghi A. A., Puliafito C. A. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age‐related macular degeneration. Ophthalmic Surg Lasers Imaging 2005; 36: 331–5
- Avery R. L., Pieramici D. J., Rabena M. D., Castellarin A. A., Nasir M. A., Giust M. J. Intravitreal bevacizumab (Avastin) for neovascular age‐related macular degeneration. Ophthalmology 2006; 113: 363–72 e5
- Vinores S. A. Anecortave (Alcon Laboratories). IDrugs 2005; 8: 327–34
- D'Amico D. J., Goldberg M. F., Hudson H., Jerdan J. A., Krueger D. S., Luna S. P., et al. Anecortave acetate as monotherapy for treatment of subfoveal neovascularization in age‐related macular degeneration: twelve‐month clinical outcomes. Ophthalmology 2003; 110: 2372–83, discussion 84–5
- Spaide R. F., Sorenson J., Maranan L. Photodynamic therapy with verteporfin combined with intravitreal injection of triamcinolone acetonide for choroidal neovascularization. Ophthalmology 2005; 112: 301–4
- Quiram P. A., Gonzales C. R., Schwartz S. D. Severe steroid‐induced glaucoma following intravitreal injection of triamcinolone acetonide. Am J Ophthalmol 2006; 141: 580–2
- Jonas J. B., Kreissig I., Spandau U. H., Harder B. Infectious and noninfectious endophthalmitis after intravitreal high‐dosage triamcinolone acetonide. Am J Ophthalmol 2006; 141: 579–80
- Guymer R. H., Chiu A. W., Lim L., Baird P. N. HMG CoA reductase inhibitors (statins): do they have a role in age‐related macular degeneration?. Surv Ophthalmol 2005; 50: 194–206
- Sarks S., Arnold J., Killingsworth M., Sarks J. Early drusen formation in the normal and aging eye and their relation to age‐related maculopathy: a clinicopathological study. Br J Ophthalmol 1999; 83: 358–68
- McConnell V., Silvestri G. Age‐related macular degeneration. Ulster Med J 2005; 74: 82–92
- Provis J. M., Penfold P. L., Cornish E. E., Sandercoe T. M., Madigan M. C. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom 2005; 88: 269–81
- Donoso L. A., Kim D., Frost A., Callahan A., Hageman G. The role of inflammation in the pathogenesis of age‐related macular degeneration. Surv Ophthalmol 2006; 51: 137–52
- Arnold J., Sarks S. Age related macular degeneration. Clin Evid 2004; 819–34
- Hageman G., Mullins R. Molecular composition of drusen as related to substructural phenotype. Mol Vis 1999; 5: 28
- Sarraf D., Gin T., Yu F., Brannon A., Owens S. L., Bird A. C. Long‐term drusen study. Retina 1999; 19: 513–9
- Klein R., Klein B., Jensen S., Meuer S. The five‐year incidence and progression of age‐related maculopathy: the Beaver Dam Eye Study. Ophthalmology 1997; 104: 7–21
- Spraul C., Grossniklaus H. Characteristics of drusen and Bruch's membrane in postmortem eyes with age‐related macular degeneration. Arch Ophthalmol 1997; 115: 267–73
- Sarks J. P., Sarks S. H., Killingsworth M. C. Evolution of soft drusen in age‐related macular degeneration. Eye 1994; 8((Pt 3))269–83
- Guymer R., Bird A. Bruch's membrane, drusen, and age‐related macular degeneration. The Retinal Pigment Epithelium, M Marmor, T Wolfensberger. Oxford University Press, New York 1998; 693–705
- Zarbin M. A. Current concepts in the pathogenesis of age‐related macular degeneration. Arch Ophthalmol 2004 Apr; 122: 598–614
- Sivaprasad S., Bailey T. A., Chong V. N. Bruch's membrane and the vascular intima: is there a common basis for age‐related changes and disease?. Clin Experiment Ophthalmol 2005; 33: 518–23
- Marshall J., Hussain A., Starita C., Moore D., Patmore A. The Retinal Pigment Epithelium. Oxford University Press, New York 1998
- Guymer R., Luthert P., Bird A. Changes in Bruch's membrane and related structures with age. Prog Retin Eye Res 1999; 18: 59–90
- Hogan M., Alvarado J. Studies on the human macula. Aging changes in Bruch's membrane. Arch Ophthalmol 1967; 77: 410–20
- Sarks S. H. Ageing and degeneration in the macular region: a clinico‐pathological study. Br J Ophthalmol 1976; 60: 324–41
- Green W. R., McDonnell P. J., Yeo J. H. Pathologic features of senile macular degeneration. Ophthalmology 1985; 92: 615–27
- Feeney‐Burns L., Ellersieck M. Age‐related changes in the ultrastructure of Bruch's membrane. Am J Ophthalmol 1985; 100: 686–97
- Grindle C., Marshall J. Ageing changes in Bruch's membrane and their functional implications. Trans Ophthal Soc UK 1978; 98: 172–5
- Pauleikhoff D., Sheraidah G., Marshall J., Bird A., Wessing A. Biochemical and histochemical analysis of age related lipid deposits in Bruch's membrane. Ophthalmologe 1994; 91: 730–4
- Handa J. T., Verzijl N., Matsunaga H., Aotaki‐Keen A., Lutty G. A., te Koppele J. M., et al. Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci 1999; 40: 775–9
- Curcio C., Millican C., Bailey T., Kruth H. Accumulation of cholesterol with age in human Bruch's membrane. Invest Ophthalmol Vis Sci 2001; 42: 265–74
- Kamei M., Hollyfield J. TIMP‐3 in Bruch's membrane: changes during aging and in age‐related macular degeneration. Invest Ophthalmol Vis Sci 1999; 40: 2367–75
- Newsome D., Huh W., Green W. Bruch's membrane age‐related changes vary by region. Curr Eye Res 1987; 6: 1211–21
- Ishibashi T., Murata T., Hangai M., Nagai R., Horiuchi S., Lopez P., et al. Advanced glycation end products in age‐related macular degeneration. Arch Ophthalmol 1998; 116: 1629–32
- Hewitt A., Nakazawa K., Newsome D. Analysis of newly synthesized Bruch's membrane proteoglycans. Invest Ophthalmol Vis Sci 1989; 30: 478–86
- Karwatowski W., Jeffries T., Duance V., Albon J., Bailey A., Easty D. Preparation of Bruch's membrane and analysis of the age‐related changes in the structural collagens. Br J Ophthalmol 1995; 79: 944–52
- Haimovici R., Gantz D. L., Rumelt S., Freddo T. F., Small D. M. The lipid composition of drusen, Bruch's membrane, and sclera by hot stage polarizing light microscopy. Invest Ophthalmol Vis Sci 2001; 42: 1592–9
- Li C. M., Chung B. H., Presley J. B., Malek G., Zhang X., Dashti N., et al. Lipoprotein‐like particles and cholesteryl esters in human Bruch's membrane: initial characterization. Invest Ophthalmol Vis Sci 2005; 46: 2576–86
- Starita C., Hussain A., Pagliarini S., Marshall J. Hydrodynamics of ageing Bruch's membrane: implications for macular disease. Exp Eye Res 1996; 62: 565–72
- Moore D., Clover G. The effect of age on the macromolecuar permeability of human Bruch's membrane. Invest Ophthalmol Vis Sci 2001; 42: 2970–5
- Hussain A., Rowe L., Marshall J. Age‐related alterations in the diffusional transport of amino acids across the human Bruch's‐choroid complex. J Opt Soc Am A Opt Image Sci Vis 2002; 19: 166–72
- Moore D., Hussain A., Marshall J. Age‐related variation in the hydraulic conductivity of Bruch's membrane. Invest Ophthalmol Vis Sci 1995; 36: 1290–7
- Chong N. H., Keonin J., Luthert P. J., Frennesson C. I., Weingeist D. M., Wolf R. L., et al. Decreased thickness and integrity of the macular elastic layer of Bruch's membrane correspond to the distribution of lesions associated with age‐related macular degeneration. Am J Pathol 2005; 166: 241–51
- Nackman G., Karkowski F., Halpern V., Gaetz H., Tilson M. Elastin degradation products induce adventitial angiogenesis in the Anidjar/Dobrin rat aneurysm model. Surgery 1997; 122: 39–44
- Kamisato S., Uemura Y., Takami N., Okamoto K. Involvement of intracellular cyclic GMP and cyclic GMP‐dependent protein kinase in alpha‐elastin‐induced macrophage chemotaxis. J Biochem (Tokyo) 1997; 121: 862–7
- Marshall J., Hussain A., Starita C., Moore D., Patmore A. Aging and Bruch's membrane. The Retinal Pigment Epithelium, M Marmor, T Wolfensberger. Oxford University Press, New York 1998; 669–92
- Holz F., Sheraiadah G., Pauleikhoff D., Marshall J., Bird A. Analysis of lipid deposits extracted from human macular and peripheral Bruch's membrane. Arch Ophthalmol 1994; 112: 402–6
- Pauleikhoff D., Harper C., Marshall J., Bird A. Aging changes in Bruch's membrane. Ophthalmology 1990; 97: 171–8
- Anderson D., Ozaki S., Nealon M., Neitz J., Mullins R., Hageman G., et al. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: Implications for the process of drusen formation. Am J Ophthalmol 2001; 131: 767–81
- Farkas T., Sylvester V., Archer D., Altona M. The histochemistry of drusen. Am J Ophthalmol 1971; 71: 1206–15
- Green W. R., Enger C. Age‐related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology 1993; 100: 1519–35
- Kliffen M., van der Schaft T. L., Mooy C. M., de Jong P. T. Morphologic changes in age‐related maculopathy. Microsc Res Tech 1997; 36: 106–22
- Feher J., Valu L. [On the structure of Bruch's membrane]. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1967; 173: 162–7
- Loffler K. U., Lee W. R. Basal linear deposit in the human macula. Graefes Arch Clin Exp Ophthalmol 1986; 224: 493–501
- van der Schaft T. L., de Bruijn W. C., Mooy C. M., Ketelaars D. A., de Jong P. T. Is basal laminar deposit unique for age‐related macular degeneration? [see comments]. Arch Ophthalmol 1991; 109: 420–5
- van der Schaft T., Mooy C., de Bruijn W., Oron F., Mulder P., de Jong P. Histological features of age‐related macular degeneration. A statistical analysis. Ophthalmology 1992; 99: 278–86
- Spraul C. W., Lang G. E., Grossniklaus H. E. Morphometric analysis of the choroid, Bruch's membrane, and retinal pigment epithelium in eyes with age‐related macular degeneration [see comments]. Invest Ophthalmol Vis Sci 1996; 37: 2724–35
- Milam A. H., Curcio C. A., Cideciyan A. V., Saxena S., John S. K., Kruth H. S., et al. Dominant late‐onset retinal degeneration with regional variation of sub‐retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology 2000; 107: 2256–66
- Kliffen M., Mooy C. M., Luider T. M., Huijmans J. G., Kerkvliet S., de Jong P. T. Identification of glycosaminoglycans in age‐related macular deposits. Arch Ophthalmol 1996; 114: 1009–14
- Kliffen M., Mooy C. M., Luider T. M., de Jong P. T. Analysis of carbohydrate structures in basal laminar deposit in aging human maculae. Invest Ophthalmol Vis Sci 1994; 35: 2901–5
- van der Schaft T., Mooy C., de Bruijn W., Bosman F., de Jong P. Immunohistochemical light and electron microscopy of basal laminar deposit. Graefes Arch Clin Exp Ophthalmol 1994; 232: 40–6
- Knupp C., Amin S., Munro P., Luthert P., Squire J. Collagen VI assemblies in age‐related macular degeneration. J Struct Biol 2002; 139: 181–9
- Curcio C. A., Presley J. B., Millican C. L., Medeiros N. E. Basal deposits and drusen in eyes with age‐related maculopathy: evidence for solid lipid particles. Exp Eye Res 2005; 80: 761–75
- Sarks S., Van Driel D., Maxwell L., Killingsworth M. Softening of drusen and subretinal neovascularization. Trans Ophthalmol Soc U K 1980; 100: 414–22
- Okubo A., Rosa R., Bunce C., Alexander R., Fan J., Bird A., et al. The relationships of age changes in retinal pigment epithelium and Bruch's membrane. Invest Ophthalmol Vis Sci 1999; 40: 443–9
- Hogan M. Role of the retinal pigment epithelium in macular disease. Trans Am Acad Ophthalmol Otolaryngol 1972; 76: 64–80
- Bird A. Pathophysiology of AMD. Age‐related macular degeneration: principles and practice, G. R Hampton, P. T Nelsen. Raven Press, New York 1992, Chapter 3
- Dorey C., Wu G., Ebenstein D. Cell loss in the aging retina: Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989; 30: 1691–9
- Bird A. C. Bruch's membrane change with age. Br J Ophthalmol 1992; 76: 166–8
- Katz M. L., Robison W. G., Jr. Age‐related changes in the retinal pigment epithelium of pigmented rats. Exp Eye Res 1984; 38: 137–51
- Feeney‐Burns L., Hilderbrand E., Eldridge S. Aging human RPE: morphometric analysis of macular, equatorial and peripheral cells. Invest Ophthalmol Vis Sci 1984; 25: 195–200
- Wing G., Blanchard G., Weiter J. Topography and age relationship of lipofuscin concentration in the RPE. Invest Ophthalmol Vis Sci 1978; 7: 601–7
- Rozanowska M., Jarvis‐Evans J., Korytowski W., Boulton M. E., Burke J. M., Sarna T. Blue light‐induced reactivity of retinal age pigment. In vitro generation of oxygen‐reactive species. J Biol Chem 1995; 270: 18825–30
- Holz F., Bellman C., Staudt S., Schutt F., Volcker H. Fundus autofluorescence and development of geographic atrophy in age‐related macular degeneration. Invest Ophthalmol Vis Sci 2001; 42: 1051–6
- Katz M. L. Potential role of retinal pigment epithelial lipofuscin accumulation in age‐related macular degeneration. Arch Gerontol Geriatr 2002; 34: 359–70
- Sparrow J. R., Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res 2005; 80: 595–606
- Ramrattan R., van der Schaft T., Mooy C., de Bruijn W., Mulder P., de Jong P. Morphometric analysis of Bruch's membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci 1994; 35: 2857–64
- McLeod D. S., Taomoto M., Otsuji T., Green W. R., Sunness J. S., Lutty G. A. Quantifying changes in RPE and choroidal vasculature in eyes with age‐related macular degeneration. Invest Ophthalmol Vis Sci 2002 Jun; 43: 1986–93
- Midena E., Segato T., Blarzino M. C., Degli Angeli C. Macular drusen and the sensitivity of the central visual field. Doc Ophthalmol 1994; 88: 179–85
- Curcio C., Medeiros N., Millican C. Photoreceptor loss in age‐related macular degeneration. Invest Ophthalmol Vis Sci 1996; 37: 1236–49
- Jackson G. R., Owsley C. Visual dysfunction, neurodegenerative diseases, and aging. Neurol Clin 2003; 21: 709–28
- Li J., Tso M. O., Lam T. T. Reduced amplitude and delayed latency in foveal response of multifocal electroretinogram in early age related macular degeneration. Br J Ophthalmol 2001; 85: 287–90
- Owsley C., Jackson G. R., Cideciyan A. V., Huang Y., Fine S. L., Ho A. C., et al. Psychophysical evidence for rod vulnerability in age‐related macular degeneration. Invest Ophthalmol Vis Sci 2000; 41: 267–73
- Jackson G. R., Owsley C., Curcio C. A. Photoreceptor degeneration and dysfunction in aging and age‐related maculopathy. Ageing Res Rev 2002; 1: 381–96
- Owsley C., Jackson G. R., White M., Feist R., Edwards D. Delays in rod‐mediated dark adaptation in early age‐related maculopathy. Ophthalmology 2001; 108: 1196–202
- Jackson G. R., McGwin G., Jr., Phillips J. M., Klein R., Owsley C. Impact of aging and age‐related maculopathy on activation of the a‐wave of the rod‐mediated electroretinogram. Invest Ophthalmol Vis Sci 2004; 45: 3271–8
- Owsley C., McGwin G., Jackson G. R., Heimburger D. C., Piyathilake C. J., Klein R., et al. Effect of short‐term, high‐dose retinol on dark adaptation in aging and early age‐related maculopathy. Invest Ophthalmol Vis Sci 2006; 47: 1310–8
- Stangos N., Voutas S., Topouzis F., Karampatakis V. Contrast sensitivity evaluation in eyes predisposed to age‐related macular degeneration and presenting normal visual acuity. Ophthalmologica 1995; 209: 194–8
- Sunness J., Massof R., Johnson M., Bressler N., Bressler S., Fine S. Diminished foveal sensitivity may predict the development of advanced age‐related macular degeneration. Ophthalmology 1989; 96: 375–81
- Kleiner R. C., Enger C., Alexander M. F., Fine S. L. Contrast sensitivity in age‐related macular degeneration. Arch Ophthalmol 1988; 106: 55–7
- Sandberg M. A., Gaudio A. R. Slow photostress recovery and disease severity in age‐related macular degeneration. Retina 1995; 15: 407–12
- Eisner A., Klein M. L., Zilis J. D., Watkins M. D. Visual function and the subsequent development of exudative age‐related macular degeneration. Invest Ophthalmol Vis Sci 1992; 33: 3091–102
- Eisner A., Stoumbos V. D., Klein M. L., Fleming S. A. Relations between fundus appearance and function. Eyes whose fellow eye has exudative age‐related macular degeneration. Invest Ophthalmol Vis Sci 1991; 32: 8–20
- Johnson P. T., Brown M. N., Pulliam B. C., Anderson D. H., Johnson L. V. Synaptic pathology, altered gene expression, and degeneration in photoreceptors impacted by drusen. Invest Ophthalmol Vis Sci 2005; 46: 4788–95
- Johnson P. T., Lewis G. P., Talaga K. C., Brown M. N., Kappel P. J., Fisher S. K., et al. Drusen‐associated degeneration in the retina. Invest Ophthalmol Vis Sci 2003; 44: 4481–8
- Hageman G. S., Luthert P. J., Victor Chong N. H., Johnson L. V., Anderson D. H., Mullins R. F. An integrated hypothesis that considers drusen as biomarkers of immune‐mediated processes at the rpe‐bruch's membrane interface in aging and age‐related macular degeneration. Prog Retin Eye Res 2001; 20: 705–32
- Anderson D., Mullins R., Hageman G., Johnson L. V. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 2002; 134: 411–31
- Killingsworth M., Sarks J., Sarks S. Macrophages related to Bruch's membrane in age‐related macular degeneration. Eye 1990; 4: 613–21
- Penfold P. L., Madigan M. C., Gillies M. C., Provis J. M. Immunological and Aetiological Aspects of Macular Degeneration. Progress in Retinal Research. Elsevier Science Ltd, London 2001; 385–414
- Heriot W., Henkind P., Bellhorn R., Burns M. Choroidal neovascularization can digest Bruch's membrane. A prior break is not essential. Ophthalmology 1984; 91: 1603–8
- Penfold P., Killingsworth M., Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch's membrane. Aust J Ophthalmol 1984; 12: 23–31
- Dastgheib K., Green W. Granulomatous reaction to Bruch's membrane in age‐related macular degeneration. Arch Ophthalmol 1994; 112: 813–8
- Hageman G., Mullins R., Russell S., Johnson L., Anderson D. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J 1999; 13: 477–84
- Mullins R. F., Russell S. R., Anderson D. H., Hageman G. S. Drusen associated with aging and age‐related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 2000; 14: 835–46
- Johnson L., Ozaki S., Staples M., Erickson P., Anderson D. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res 2000; 70: 441–9
- Johnson L., Leitner W., Staples M., Anderson D. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res 2001; 73: 887–96
- Mullins R. F., Johnson L. V., Anderson D. H., Hageman G. S. Characterization of drusen‐associated glycoconjugates. Ophthalmology 1997; 104: 288–94
- Russell S. R., Mullins R. F., Schneider B. L., Hageman G. S. Location, substructure, and composition of basal laminar drusen compared with drusen associated with aging and age‐related macular degeneration. Am J Ophthalmol 2000; 129: 205–14
- Mullins R. F., Aptsiauri N., Hageman G. S. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye 2001; 15((Pt 3))390–5
- Johnson L. V., Leitner W. P., Rivest A. J., Staples M. K., Radeke M. J., Anderson D. H. The Alzheimer's A beta‐peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age‐related macular degeneration. Proc Natl Acad Sci U S A 2002; 99: 11830–5
- Malek G., Li C. M., Guidry C., Medeiros N. E., Curcio C. A. Apolipoprotein B in cholesterol‐containing drusen and basal deposits of human eyes with age‐related maculopathy. Am J Pathol 2003; 162: 413–25
- Anderson D., Talaga K., Rivest A., Barron E., Hageman G., Johnson L. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age‐related macular degeneration. Exp Eye Res 2004; 78: 243–56
- Peto T., Lengyel I., Frederickson C., Vankuijk F. Zinc in Drusen. Invest Ophthalmol Vis Sci 2005; 46, E‐Abstract 3008
- Holers V. M. The complement system as a therapeutic target in autoimmunity. Clin Immunol 2003; 107: 140–51
- Fearon D. T., Locksley R. M. The instructive role of innate immunity in the acquired immune response. Science 1996; 272: 50–3
- Mastellos D., Andronis C., Persidis A., Lambris J. D. Novel biological networks modulated by complement. Clin Immunol 2005; 115: 225–35
- Mastellos D., Germenis A. E., Lambris J. D. Complement: an inflammatory pathway fulfilling multiple roles at the interface of innate immunity and development. Curr Drug Targets Inflamm Allergy 2005; 4): 125–7
- Walport M. J. Complement. Second of two parts. N Engl J Med 2001; 344: 1140–4
- Walport M. J. Complement. First of two parts. N Engl J Med 2001; 344: 1058–66
- Thurman J. M., Holers V. M. The central role of the alternative complement pathway in human disease. J Immunol 2006; 176: 1305–10
- Pio R., Martinez A., Unsworth E. J., Kowalak J. A., Bengoechea J. A., Zipfel P. F., et al. Complement factor H is a serum‐binding protein for adrenomedullin, and the resulting complex modulates the bioactivities of both partners. J Biol Chem 2001; 276: 12292–300
- Holers V. M., Thurman J. M. The alternative pathway of complement in disease: opportunities for therapeutic targeting. Mol Immunol 2004; 41: 147–52
- Song W. C. Membrane complement regulatory proteins in autoimmune and inflammatory tissue injury. Curr Dir Autoimmun 2004; 7: 181–99
- Zipfel P. F., Heinen S., Jozsi M., Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol 2006; 43: 97–106
- Halstensen T. S., Mollnes T. E., Fausa O., Brandtzaeg P. Deposits of terminal complement complex (TCC) in muscularis mucosae and submucosal vessels in ulcerative colitis and Crohn's disease of the colon. Gut 1989; 30: 361–6
- Sospedra M., Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005; 23: 683–747
- Lassmann H. Recent neuropathological findings in MS—implications for diagnosis and therapy. J Neurol 2004 Sep; 251(Suppl 4)IV2–5
- Neumann E., Barnum S. R., Tarner I. H., Echols J., Fleck M., Judex M., et al. Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum 2002; 46: 934–45
- Tang S., Lui S. L., Lai K. N. Pathogenesis of lupus nephritis: an update. Nephrology (Carlton) 2005; 10: 174–9
- Turnberg D., Cook H. T. Complement and glomerulonephritis: new insights. Curr Opin Nephrol Hypertens 2005; 14: 223–8
- Gerard N. P., Gerard C. Complement in allergy and asthma. Curr Opin Immunol 2002; 14: 705–8
- Rodriguez de Cordoba S., Esparza‐Gordillo J., Goicoechea de Jorge E., Lopez‐Trascasa M., Sanchez‐Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol 2004; 41: 355–67
- Pangburn M. K., Pangburn K. L., Koistinen V., Meri S., Sharma A. K. Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J Immunol 2000; 164: 4742–51
- Zipfel P. F. Complement factor H: physiology and pathophysiology. Semin Thromb Hemost 2001; 27: 191–9
- Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci U S A 2005; 102: 7227–32
- Johnson L. V., Talaga K. C., Rivest A. J., Staples M. K., Radeke M. J., Barron E., et al. Characterization of Beta Amyloid‐Containing Vesicles within Drusen. Invest Ophthalmol Vis Sci 2003; 44, E‐Abstract 4229
- Hageman G., Luthert P., Chong N., Johnson L., Anderson D., Mullins R. An integrated hypothesis that considers drusen as biomarkers of immune‐mediated processes at the RPE‐Bruch's membrane interface in aging and age‐related macular degeneration. Prog Retin Eye Res 2001; 20: 705–32
- Miller D. M., Espinosa‐Heidmann D. G., Legra J., Dubovy S. R., Suner I. J., Sedmak D. D., et al. The association of prior cytomegalovirus infection with neovascular age‐related macular degeneration. Am J Ophthalmol 2004; 138: 323–8
- Klein M., Mauldin W., Stoumbos V. Heredity and age‐related macular degeneration. Observations in monozygotic twins. Arch Ophthalmol 1994; 112: 932–7
- Heiba I., Elston R., Klein B., Klein R. Sibling correlations and segregation analysis of age‐related maculopathy: the Beaver Dam Eye Study. Genet Epidemiol 1994; 11: 51–67
- Seddon J., Ajani U., Mitchell B. Familial aggregation of age‐related maculopathy. Am J Ophthalmol 1997; 123: 199–206
- Meyers S. M., Greene T., Gutman F. A. A twin study of age‐related macular degeneration. Am J Ophthalmol 1995; 120: 757–66
- Hammond C. J., Webster A. R., Snieder H., Bird A. C., Gilbert C. E., Spector T. D. Genetic influence on early age‐related maculopathy: a twin study. Ophthalmology 2002; 109: 730–6
- Abecasis G. R., Yashar B. M., Zhao Y., Ghiasvand N. M., Zareparsi S., Branham K. E., et al. Age‐related macular degeneration: a high‐resolution genome scan for susceptibility loci in a population enriched for late‐stage disease. Am J Hum Genet 2004; 74: 482–94
- Iyengar S. K., Song D., Klein B. E., Klein R., Schick J. H., Humphrey J., et al. Dissection of genomewide‐scan data in extended families reveals a major locus and oligogenic susceptibility for age‐related macular degeneration. Am J Hum Genet 2004; 74: 20–39
- Klein M. L., Schultz D. W., Edwards A., Matise T. C., Rust K., Berselli C. B., et al. Age‐related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol 1998; 116: 1082–8
- Majewski J., Schultz D. W., Weleber R. G., Schain M. B., Edwards A. O., Matise T. C., et al. Age‐related macular degeneration—a genome scan in extended families. Am J Hum Genet 2003; 73: 540–50
- Schick J. H., Iyengar S. K., Klein B. E., Klein R., Reading K., Liptak R., et al. A whole‐genome screen of a quantitative trait of age‐related maculopathy in sibships from the Beaver Dam Eye Study. Am J Hum Genet 2003; 72: 1412–24
- Seddon J. M., Santangelo S. L., Book K., Chong S., Cote J. A genomewide scan for age‐related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet 2003; 73: 780–90
- Weeks D. E., Conley Y. P., Mah T. S., Paul T. O., Morse L., Ngo‐Chang J., et al. A full genome scan for age‐related maculopathy. Hum Mol Genet 2000; 9: 1329–49
- Weeks D. E., Conley Y. P., Tsai H. J., Mah T. S., Rosenfeld P. J., Paul T. O., et al. Age‐related maculopathy: an expanded genome‐wide scan with evidence of susceptibility loci within the 1q31 and 17q25 regions. Am J Ophthalmol 2001; 132: 682–92
- Weeks D. E., Conley Y. P., Tsai H. J., Mah T. S., Schmidt S., Postel E. A., et al. Age‐Related Maculopathy: A Genomewide Scan with Continued Evidence of Susceptibility Loci within the 1q31, 10q26, and 17q25 Regions. Am J Hum Genet 2004; 75((2))174–89
- Fisher S. A., Abecasis G. R., Yashar B. M., Zareparsi S., Swaroop A., Iyengar S. K., et al. Meta‐analysis of genome scans of age‐related macular degeneration. Hum Mol Genet 2005; 14: 2257–64
- Schultz D., Klein M., Humpert A., Luzier C., Persun V., Schain M., et al. Analysis of the ARMD1 locus: Evidence that a mutation in HEMICENTIN‐1 is associated with age‐related macular degeneration in a large family. Hum Mol Genet 2003; 12: 3315–23
- Conley Y. P., Thalamuthu A., Jakobsdottir J., Weeks D. E., Mah T., Ferrell R. E., et al. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age‐related maculopathy. Hum Mol Genet 2005; 14: 1991–2002
- Haines J. L., Schnetz‐Boutaud N., Schmidt S., Scott W. K., Agarwal A., Postel E. A., et al. Functional candidate genes in age‐related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci 2006; 47: 329–35
- Stone E. M., Braun T. A., Russell S. R., Kuehn M. H., Lotery A. J., Moore P. A., et al. Missense variations in the fibulin 5 gene and age‐related macular degeneration. N Engl J Med 2004; 351: 346–53
- Zareparsi S., Buraczynska M., Branham K. E., Shah S., Eng D., Li M., et al. Toll‐like receptor 4 variant D299G is associated with susceptibility to age‐related macular degeneration. Hum Mol Genet 2005; 14: 1449–55
- Souied E. H., Benlian P., Amouyel P., Feingold J., Lagarde J. P., Munnich A., et al. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age‐related macular degeneration. Am J Ophthalmol 1998; 125: 353–9
- Klaver C. C., Kliffen M., van Duijn C. M., Hofman A., Cruts M., Grobbee D. E., et al. Genetic association of apolipoprotein E with age‐related macular degeneration. Am J Hum Genet 1998; 63: 200–6
- Baird P. N., Guida E., Chu D. T., Vu H. T., Guymer R. H. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age‐related macular degeneration. Invest Ophthalmol Vis Sci 2004; 45: 1311–5
- Allikmets R., Shroyer N., Singh N., Seddon J., Lewis R., Bernstein P., et al. Mutation of the Stargardt disease gene (ABCR) in age‐related macular degeneration. Science 1997; 277: 1805–7
- Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., et al. Complement factor H polymorphism in age‐related macular degeneration. Science 2005; 15(308)385–9
- Edwards A. O., Ritter R., 3rd., Abel K. J., Manning A., Panhuysen C., Farrer L. A. Complement factor H polymorphism and age‐related macular degeneration. Science 2005; 308: 421–4
- Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., et al. Complement factor H variant increases the risk of age‐related macular degeneration. Science 2005; 308: 419–21
- Zareparsi S., Branham K. E., Li M., Shah S., Klein R. J., Ott J., et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age‐related macular degeneration. Am J Hum Genet 2005; 77: 149–53
- Magnusson K. P., Duan S., Sigurdsson H., Petursson H., Yang Z., Zhao Y., et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med 2006; 3: e5
- Sepp T., Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Moore A. T., et al. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Invest Ophthalmol Vis Sci 2006; 47: 536–40
- Souied E. H., Leveziel N., Richard F., Dragon‐Durey M. A., Coscas G., Soubrane G., et al. Y402H complement factor H polymorphism associated with exudative age‐related macular degeneration in the French population. Mol Vis 2005; 11: 1135–40
- Okamoto H., Umeda S., Obazawa M., Minami M., Noda T., Mizota A., et al. Complement factor H polymorphisms in Japanese population with age‐related macular degeneration. Mol Vis 2006; 12: 156–8
- Gold B., Merriam J. E., Zernant J., Hancox L. S., Taiber A. J., Gehrs K., et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age‐related macular degeneration. Nat Genet 2006; 38: 458–62
- Lokki M. L., Koskimies S. A. Allelic differences in hemolytic activity and protein concentration of BF molecules are found in association with particular HLA haplotypes. Immunogenetics 1991; 34: 242–6
- Jakobsdottir J., Conley Y. P., Weeks D. E., Mah T. S., Ferrell R. E., Gorin M. B. Susceptibility genes for age‐related maculopathy on chromosome 10q26. Am J Hum Genet 2005; 77: 389–407
- Rivera A., Fisher S. A., Fritsche L. G., Keilhauer C. N., Lichtner P., Meitinger T., et al. Hypothetical LOC387715 is a second major susceptibility gene for age‐related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet 2005; 14: 3227–36
- Schmidt S., Hauser M. A., Scott W. K., Postel E. A., Agarwal A., Gallins P., et al. Cigarette smoking strongly modifies the association of LOC387715 and age‐related macular degeneration. Am J Hum Genet 2006; 78: 852–64
- Saunders R. E., Goodship T. H., Zipfel P. F., Perkins S. J. An interactive web database of factor H‐associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease‐associated mutations. Hum Mutat 2006; 27: 21–30
- Heinen S., Sanchez‐Corral P., Jackson M. S., Strain L., Goodship J. A., Kemp E. J., et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat 2006; 27: 292–3
- Warwicker P., Goodship T. H., Donne R. L., Pirson Y., Nicholls A., Ward R. M., et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int 1998; 53: 836–44
- Esparza‐Gordillo J., Goicoechea de Jorge E., Buil A., Carreras Berges L., Lopez‐Trascasa M., Sanchez‐Corral P., et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet 2005; 14: 703–12
- Caprioli J., Castelletti F., Bucchioni S., Bettinaglio P., Bresin E., Pianetti G., et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C‐257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet 2003; 12: 3385–95
- Caprioli J., Noris M., Brioschi S., Pianetti G., Castelletti F., Bettinaglio P., et al. Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006; 108: 1267–79
- Atkinson J. P., Liszewski M. K., Richards A., Kavanagh D., Moulton E. A. Hemolytic uremic syndrome: an example of insufficient complement regulation on self‐tissue. Ann N Y Acad Sci 2005; 1056: 144–52
- Abrera‐Abeleda M. A., Nishimura C., Smith J. L., Sethi S., McRae J. L., Murphy B. F., et al. Variations in the Complement Regulatory Genes Factor H (CFH) and Factor H Related 5 (CFHR5) are Associated with Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease). J Med Genet 2006; 43: 582–9
- Appel G. B., Cook H. T., Hageman G., Jennette J. C., Kashgarian M., Kirschfink M., et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol 2005; 16: 1392–403
- Robman L., Mahdi O., McCarty C., Dimitrov P., Tikellis G., McNeil J., et al. Exposure to Chlamydia pneumoniae infection and progression of age‐related macular degeneration. Am J Epidemiol 2005; 161: 1013–9
- Erkkila H., Raitta C., Niemi K. Ocular findings in four siblings with pseudoxanthoma elasticum. Acta Ophthalmol (Copenh) 1983; 61: 589–99
- Zee R. Y., Diehl K. A., Ridker P. M. Complement factor H Y402H gene polymorphism, C‐reactive protein, and risk of incident myocardial infarction, ischaemic stroke, and venous thromboembolism: A nested case‐control study. Atherosclerosis 2006; 187: 332–5