Abstract
The anterior pituitary gland is a central regulator of growth, reproduction and homeostasis, and is the end‐product of a carefully orchestrated pattern of expression of signalling molecules and transcription factors leading to the development of this complex organ secreting six hormones from five different cell types. Naturally occurring and transgenic murine models have demonstrated a role for many of these molecules in the aetiology of combined pituitary hormone deficiency (CPHD). These include the transcription factors HESX1, PROP1, POU1F1, LHX3, LHX4, TBX19 (TPIT), SOX3 and SOX2. The expression pattern of these transcription factors, their interaction with co‐factors and their impact on target genes dictate the phenotype that results when the gene encoding the relevant transcription factor is mutated. The highly variable phenotype may consist of isolated hypopituitarism, or more complex disorders such as septo‐optic dysplasia (SOD) and holoprosencephaly. Since mutations in any one transcription factor are uncommon, and since the overall incidence of mutations in known transcription factors is low in patients with CPHD, it is clear that many genes remain to be identified, and characterization of these will further elucidate the pathogenesis of these complex conditions, and also shed light on normal pituitary development.
Hypothalamo‐pituitary development
The pituitary gland is a central regulator of growth, reproduction and homeostasis, mediated via hormone‐signalling pathways which act to regulate the finely balanced homeostatic control in vertebrates. These pathways co‐ordinate complex signals from the brain and the hypothalamus to various target organs such as the adrenals, thyroid and gonads. The pituitary is situated within the sella turcica at the base of the brain, and the mature gland consists of the adenohypophysis (anterior and intermediate lobes) and the neurohypophysis (posterior lobe), which have distinct developmental origins. Hormones secreted from the anterior pituitary regulate growth, puberty, metabolism, response to stress, reproduction and lactation. These hormones are produced from five different cell types comprising the anterior pituitary: somatotropes (growth hormone, GH), thyrotropes (thyrotropin or thyroid stimulating hormone, TSH), corticotropes (corticotropin or adrenocorticotropic hormone, ACTH), gonadotropes (follicle‐stimulating hormone, FSH, and luteinizing hormone, LH) and lactotropes (prolactin). The intermediate lobe contains melanotropes, the sixth hormone producing cell type in the pituitary, producing pro‐opiomelanocortin (POMC), which is the precursor to melanocyte‐stimulating hormone (MSH) in addition to endorphins. POMC is also the precursor to ACTH and is additionally expressed by corticotropes in the anterior lobe. The posterior lobe contains the axonal projections of neurons which are resident in the hypothalamus, and releases oxytocin, required during parturition and lactation, and arginine vasopressin (AVP), involved in regulation of osmotic balance, under the control of the hypothalamus.
The hypothalamus lies in a superior position to the mature pituitary gland and has neural projections into the cerebral cortex and the median eminence. Hypothalamic hormones are transported to the intermediate and anterior lobes of the pituitary via the portal blood system. Both the neural axonal projections to the pituitary gland and the portal blood supply are carried by the infundibulum or pituitary stalk. Therefore, any damage to the pituitary stalk can result in both anterior and posterior pituitary dysfunction.
The pituitary is derived from the midline of the anterior neural ridge and the development of the pituitary gland is similar in all vertebrates studied with the three lobes of the mature gland having a dual embryonic origin. The anterior and intermediate lobes of the pituitary are derived from the oral ectoderm, whilst the posterior pituitary originates from neural ectoderm. Development of the anterior pituitary occurs in distinct stages of differentiation and has been extensively studied in the mouse. Initiating at 7.5 days post‐coitum (dpc) in the mouse, the hypophyseal or pituitary placode is derived from thickening of the ectoderm at the roof of the primitive oral cavity that makes contact with the floor of the ventral diencephalon. Next, at approximately 9 dpc, the oral ectoderm invaginates to form a rudimentary Rathke's pouch, the primordium of the anterior pituitary, whilst the ventral diencephalon evaginates to form the infundibulum and posterior pituitary. Thirdly, a definitive Rathke's pouch is formed, finally followed by the spatial and temporal differentiation of the various cell types within the mature anterior pituitary gland. At approximately 12.5 dpc, differentiated corticotropes appear in the ventral region of the pouch and thyrotropes a day later. This is followed by the differentiation of melanotropes in the intermediate lobe at 14.5 days, and somatotrope, lactotrope and gonadotrope cells arise temporally between 15.5–16 days. By 17.5 dpc all of the hormone‐secreting cell types have undergone terminal differentiation and are organized into distinct spatial networks within the gland.
The juxtaposition of the oral ectoderm forming Rathke's pouch and the neural ectoderm of the diencephalon is maintained throughout the early stages of pituitary organogenesis, and inductive tissue interactions and extrinsic signalling from the neuroectoderm of the infundibulum are critical for the initial development of the pituitary gland Citation1. A cascade of signalling molecules and transcription factors play crucial roles in organ commitment, cell proliferation, cell patterning and terminal differentiation events within the developing pituitary (Figure ). Extrinsic signalling molecules expressed from the infundibular region of the ventral diencephalon (bone morphogenetic protein 4 (Bmp 4), fibroblast growth factors 8 and 10 (Fgf8, Fgf10), thyroid transcription factor (Ttf1; also called Nkx2.1)) together with signals expressed from the oral ectoderm (Sonic Hedgehog (Shh)) and signals from within the pouch itself (Bmp2, and Wnt4) play critical roles in early pituitary organogenesis (extensively reviewed elsewhere Citation2–4).
Figure 1 Cascade of signalling molecules and transcription factors in the developing pituitary.
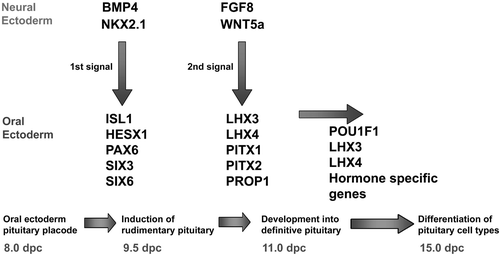
In comparison with the rodent, relatively little is known about pituitary development in humans. However, it would appear that several transcription factors involved in the embryological development of the murine pituitary may also be involved in human pituitary organogenesis. Spontaneous or artificially induced mutations and gene knockouts in the mouse have led to significant insights into human pituitary disease, with the identification of human mutations in a number of genes which give rise to hypopituitary phenotypes in their respective murine orthologues. Within Rathke's pouch and the developing anterior pituitary, a number of transcription factors involved in patterning and differentiation of the different hormone‐producing cell lineages are expressed in a spatially and temporally specific manner. Many have been implicated in the aetiology of both murine and human hypopituitarism including Hesx1, Lhx3, Lhx4, Prophet of Pit1 (Prop1), Pou1f1 (previously called Pit‐1), Pitx2 and T‐Pit (Tbx19) (Table ) Citation5. This review will deal primarily with transcription factors implicated in the aetiology of hypopituitarism in humans as a result of the identification and characterization of mutations within these genes in patients and their respective murine orthologues.
Table I. Human mutations causing abnormal hypothalamo‐pituitary development and function.
Key messages
Normal anterior pituitary development is dependent upon a complex genetic cascade of transcription factors and signalling molecules.
Mutations in HESX1, SOX2, SOX3, LHX3, LHX4, PROP1, POU1F1, and TBX19/TPIT have been associated with septo optic dysplasia/combined pituitary hormone deficiency/isolated growth hormone deficiency or isolated adrenocorticotropic hormone deficiency.
The phenotypes are dependent on the expression patterns of the genes, and probably the interactions with co‐factors, and the impact on target genes.
The inheritance and penetrance can be variable.
Hypopituitarism
Congenital hypopituitarism encompasses a group of different aetiological disorders, and may manifest as isolated hormonal deficiencies (isolated growth hormone deficiency, IGHD; ACTH deficiency; gonadotropin deficiency (hypogonadotropic hypogonadism); TSH deficiency) or as combined pituitary hormone deficiency (CPHD), either in isolation or associated with extra‐pituitary features such as optic nerve hypoplasia and midline forebrain defects. Clinical features of hypopituitarism are often variable, and onset may occur early in the neonatal period, often with a stormy perinatal course and/or conjugated hyperbilirubinaemia, or later with growth failure or abnormal pubertal development. Isolated GH deficiency is by far the most common endocrinopathy and may present with growth failure and consequent short stature. The evolution of additional hormone deficiencies with time is now a well recognized feature of hypopituitarism, and hence a complete evaluation of the hypothalamo‐pituitary axis is indicated in any patient suspected of having one hormonal deficiency. Investigations include the use of combined pituitary function testing, measurement of insulin‐like growth factor I and its binding protein (IGF‐1 and IGFBP3), and neuroimaging Citation6. Hormonal replacement is the mainstay of treatment, and entails the use of subcutaneously administered recombinant human growth hormone (r‐hGH), oral hydrocortisone and/or thyroxine and intramuscular or transdermal testosterone or oestrogen.
The incidence of congenital IGHD, the most common congenital manifestation of hypopituitarism, varies from 1 in 3500 to 1 in 10,000 Citation7–11, with the majority of cases being idiopathic in origin. However, familial inheritance, which can be dominant or recessive, accounts for 5%–30% of cases Citation12, suggesting a genetic aetiology for the condition. Septo‐optic dysplasia (SOD) is a rare congenital anomaly with a relative prevalence of 1 per 10,000 Citation13. It is a heterogeneous disorder characterized by a clinical triad of midline forebrain abnormalities, optic nerve hypoplasia (ONH) and hypopituitarism. Although hypopituitarism is more common in males (M:F ratio 2:1), SOD is thought to be equally prevalent in males and females and, although the condition is generally sporadic, familial cases have been described. The phenotype is highly variable, and each of the three components can occur in isolation or in combination with at least one of the other components. The precise aetiology of the condition still remains unknown and is most likely to be multifactorial, with a combination of genetic and environmental factors Citation13, Citation14.
Over the last 35 years, significant advances in molecular biology and our understanding of the normal development of the pituitary gland have led to a greater understanding of the genetic basis of hypopituitarism. Spontaneously occurring and transgenic murine models have shed considerable light on the aetiology of congenital hypopituitarism in humans.
Transcription factors associated with hypopituitarism
HESX1
Hesx1 (also referred to as Rathke's pouch homeobox, RpxCitation15) is a member of the paired‐like class of homeobox transcription factors initially isolated from undifferentiated embryonic stem cells Citation16. Hesx1 functions as a transcriptional repressor, with repression domains within the N terminal region and the DNA binding homeodomain Citation17, Citation18, and is one of the earliest markers of the murine pituitary primordium, initially expressed during gastrulation in a very restricted region of anterior endoderm. At later stages of development, Hesx1 is expressed in a region fated to form the forebrain and ventral diencephalon, followed by resolution to Rathke's pouch by 9–9.5 dpc. Thereafter expression is limited to the pouch only and the subsequent developing anterior pituitary from 9.5–13.5 dpc, after which time Hesx1 expression becomes undetectable Citation15, Citation19. Experiments using transgenic mice have shown that regulatory sequences upstream of the coding region are important for early expression of the gene in the anterior endoderm and neural plate during gastrulation. These regions contain binding sites for the LIM (named after Lin‐11, Isl‐1 and Mec‐3 genes) homeodomain proteins Lhx1 and Lhx3 which are required for transcriptional activity. Moreover, conserved regulatory elements in the 3′ downstream region of Hesx1 are necessary and sufficient for later expression in the developing Rathke's pouch Citation20.
Mice with a homozygous targeted disruption of Hesx1 display morphological defects of prospective forebrain tissue and absence of developing optic vesicles. The phenotype of Hesx1 null mice is highly variable, and often asymmetric, with the most severely affected embryos exhibiting substantial reduction of the forebrain and no telencephalic vesicle or infundibular development, in addition to absence of the optic cups and Rathke's pouch Citation21. At birth, the most severely affected mutants showed a phenotype comprising markedly decreased head size, short nose and anophthalmia or severe microphthalmia with additional defects including hypoplasia of the nasal cavities and olfactory bulbs and abnormalities of the corpus callosum and septum pellucidum. A small percentage (5%) of the most severely affected neonates had complete agenesis of the anterior pituitary gland Citation17. A second class of homozygous mutants showed a milder phenotype with an overall reduction of forebrain epithelium and ventral midline defects of the hypothalamus and aberrant morphogenesis of Rathke's pouch which displayed abnormal bifurcations. The majority of these mice showed multiple invaginations of the oral ectoderm, resulting in the apparent formation of multiple pituitary glands. The olfactory pits and optic vesicles were also hypoplastic. Although exhibiting a variable phenotypic spectrum, all homozygous mutant mice displayed an abnormal phenotype at birth. However, approximately 1% of the heterozygous mice also displayed a mild phenotype, and the reason for this variability remains unknown Citation21.
The highly variable phenotype of forebrain midline defects, ocular abnormalities and pituitary dysplasia observed in Hesx1 null mice is similar to that of SOD. Subsequently, mutations in HESX1 (MIM 601802) have been identified in a small proportion of SOD patients. To date, five homozygous mutations have been reported in HESX1 associated with variable phenotypes. Dattani et al. Citation21 initially described an arginine‐to‐cysteine substitution (R160C) in two siblings descended from consanguineous parents with a severe SOD phenotype comprising panhypopituitarism with hypoplasia of the anterior pituitary and an ectopic/undescended posterior lobe, agenesis of the corpus callosum with an absent septum pellucidum, and optic nerve hypoplasia with small optic chiasma (Figure ) Citation21–23. This mutation occurs at a highly conserved residue of the homeodomain resulting in complete ablation of DNA binding. A second homozygous mutation (I26T) was identified in a girl presenting with GH and gonadotropin deficiency, with evolving ACTH and TSH deficiency. Magnetic resonance imaging (MRI) revealed anterior pituitary hypoplasia with an ectopic posterior pituitary, but with normal optic nerves and midline forebrain structures. This mutation lies in a highly conserved region in the N terminus of HESX1, the engrailed homology domain (eh‐1) crucial for transcriptional repression Citation22, and was associated with partial loss of repression Citation24 due to impaired interaction with the co‐repressor TLE1 (OMIM 600189). A third recessive mutation was identified in two siblings from a consanguineous family, involving an Alu‐element insertion in exon 3 of HESX1, which encodes the homeodomain of the protein Citation25. Homozygosity for the mutation was associated with a severe phenotype including undetectable levels of all anterior pituitary hormones as a result of aplasia of the anterior pituitary (as observed in 5% of homozygous null mice), together with hypoplasia of the sella turcica. Conversely, the posterior pituitary and pituitary stalk were both normal. One sibling had a coloboma of the right optic nerve resulting in unilateral blindness, whilst the other had no ophthalmic abnormalities but displayed a left‐sided diaphragmatic hernia and aortic coarctation and died shortly after birth. More recently, Sobrier et al. (2006) reported two additional patients with novel recessive mutations in HESX1 associated with anterior pituitary aplasia in the absence of an ectopic posterior pituitary or optic nerve abnormalities, features typically associated with HESX1 mutations. Sequencing of HESX1 in the two individuals revealed that one patient carried a homozygous 2‐bp deletion (c.449_450delAC) resulting in a frameshift, whilst the other patient was homozygous for a mutation in the splice donor site in intron 2 (c.357+2T>C), with both mutations predicted to disrupt the homeodomain of the protein Citation26. Anterior pituitary agenesis has been reported very rarely. Arrigo et al. (2006) recently described 4 patients with anterior pituitary aplasia, but failed to identify any mutations within HESX1Citation27, suggesting that mutations in other genes are likely to contribute to this rare condition.
Figure 2 A: Sagittal magnetic resonance imaging (MRI) scan of the head of a normal child. Note the well formed corpus callosum (CC) and the optic chiasm (OC) and the posterior pituitary (PP) which appears as a bright fossa within the sella turcica. B: Sagittal MRI scan of two siblings with a homozygous R160C mutation in HESX1. In the first sibling (i) the splenium of the corpus callosum is more hypoplastic than the rest of the structure and the posterior pituitary is partially descended as compared with sibling 2 (ii) who has a severely hypoplastic corpus callosum, ectopic posterior pituitary and lack of visible pituitary stalk (PS). C: Sequential MRI scanning of a patient with a 13‐bp deletion (del112‐124) in PROP1 reveals waxing and waning of a pituitary mass (denoted by arrow); (i) on presentation, (ii) 4 months after initial MRI, (iii) 12 months after initial MRI, (iv) 21 months after initial MRI. D: Coronal and sagittal MRI scans from one patient (panels (i) and (ii)) and sagittal scan from a second patient (iii) with SOX3 duplication showing anterior pituitary (AP) hypoplasia; partial hypoplasia of the infundibulum (I) in the first patient, which is absent in the second; and an ectopic/undescended posterior pituitary, more severe in patient 2. Note the cyst in the corpus callosum identified in patient 1. Figures A and B derived from Brickman et al. (22). Figures C and D derived from Turton et al. (39) and Woods et al. (108) respectively and reproduced with permission from Clinical Endocrinology (Blackwell Publishing) and the American Journal of Human Genetics (University of Chicago Press).
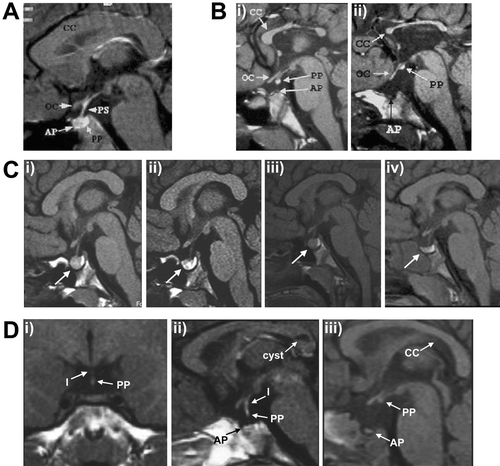
Consistent with the rare (approximately 1%) heterozygous murine phenotype Citation21, five additional heterozygous mutations of HESX1 have been identified in individuals with various forms of hypopituitarism and SOD. Thomas et al. Citation28 identified three non‐conservative amino acid substitutions in a screen of 228 patients with varying degrees of hypopituitarism, including 105 patients with SOD. These included a serine‐to‐leucine substitution at codon 170 (S170L) in two siblings with isolated GH deficiency and optic nerve hypoplasia. MRI of both patients showed a normal anterior pituitary with an ectopic posterior lobe, in association with partial agenesis of the corpus callosum, the extent of which differed between the two siblings. A threonine‐to‐alanine substitution at codon 181 (T181A) was identified in a patient with isolated GH deficiency, and a glutamine‐to‐histidine change in exon 1 (Q6H) was associated with multiple hormone deficiencies (GH, TSH and gonadotropins). Furthermore, a deletion at nucleotide position 1684 (g.1684delG), leading to a frameshift altering the C terminal portion of the protein Citation29, and a de novo 2‐bp insertion (c.306_307insAG) Citation30 have been identified in patients presenting with SOD phenotypes characterized by GH deficiency, hypoplasia of the anterior pituitary and optic nerves and midline forebrain abnormalities. The patient with the insertion mutation also exhibited evidence of gonadotropin deficiency and hypothyroidism Citation30. These heterozygous HESX1 mutations are, in general, associated with milder phenotypes than their recessive counterparts and haplotype analysis in familial cases has revealed that such mutations are associated with incomplete penetrance, with most affected individuals inheriting the mutation from one of their unaffected parents Citation28. Additionally, the T181A mutation was identified in an unaffected sibling consistent with the finding that heterozygous Hesx1+/− mice display low penetrance of a mild phenotype. Therefore, both homozygous and heterozygous mutations in HESX1 are associated with highly variable pituitary/forebrain phenotypes, and as yet, no clear genotype‐phenotype correlations have been identified.
PROP1
Prop1 is a paired‐like homeodomain transcription factor, expressed exclusively within the embryonic pituitary. Expression is first detected in the dorsal portion of the murine Rathke's pouch in a region overlapping the expression domain of Hesx1. Maximal expression of Prop1 is achieved at 12 dpc in the full caudomedial area of the developing anterior pituitary, followed by a marked decrease, becoming undetectable by 15.5 dpc Citation18. The Ames dwarf (df) mouse harbours a naturally occurring serine‐to‐proline (S83P) substitution within the homeodomain of Prop1, resulting in a mutant protein with an eight‐fold lower DNA binding affinity than wild type Prop1 Citation18. Homozygous Ames dwarf mice exhibit severe proportional dwarfism, hypothyroidism and infertility, and although early pituitary development is similar to wild type mice, the emerging anterior pituitary gland is reduced by about 50%, displaying an abnormal looping appearance Citation31. Rathke's pouch and the developing anterior gland of these mice is poorly vascularized and shows decreased proliferation and increased apoptosis postnatally, resulting in a profoundly hypoplastic anterior pituitary gland Citation32, Citation33. The adult Ames dwarf mouse exhibits GH, TSH and prolactin deficiency resulting from a severe reduction of somatotrope, lactotrope and caudomedial thyrotrope lineages with approximately 1% of the normal complement of each cell type. Additionally these mice have reduced gonadotropin expression correlating with low plasma LH and FSH levels Citation18, Citation31, Citation34. Mice homozygous for a complete deletion of Prop1 show a nearly identical phenotype to the Ames mutants when compared on the same mouse strain Citation35. Prop1 null mutants exhibit a lack of GH‐ and TSH‐positive cells and delayed gonadotrope development with reduced FSH and absent LH. However, approximately half of Prop1 null mice die of respiratory distress syndrome shortly after birth dependent on genetic background, suggesting that genetic modifiers influence the phenotype associated with Prop1 mutations Citation35.
Following the identification of Prop1 as the gene underlying the Ames dwarf phenotype in mice, Wu et al. Citation36 reported the first mutations in PROP1 (OMIM 601538) in human patients with GH, TSH and PRL deficiency in addition to reduced gonadotropins and a failure to enter puberty spontaneously. To date, 22 distinct mutations have been identified in more than 170 patients from 21 different countries, suggesting that PROP1 mutations are the most common cause of CPHD reported, accounting for approximately 50% of familial cases Citation37, Citation38, although the incidence in sporadic cases is much lower Citation38, Citation39. All affected individuals exhibit recessive inheritance, and most mutations identified to date involve the DNA binding homeodomain which is highly conserved between mouse and human sharing 91% identity at the nucleotide level Citation36, Citation40. These include three nonsense, Citation41–43, nine missense Citation36, Citation40, Citation43–48, seven frameshift mutations Citation36, Citation41, Citation45, Citation49–53, and two splice site mutations which disrupt normal exon splicing Citation40, Citation54, in addition to a complete deletion of the gene Citation55 (Table ). The majority of these mutations are predicted to result in complete loss of function of the protein by ablating DNA binding and transcriptional activation. However, in vitro analysis has shown that some missense mutations retain partial activity Citation36, Citation44, Citation56. By far the most common mutation (50%–72% of all familial PROP1 mutations Citation37, Citation38, Citation45), detected in multiple unrelated families from several different countries, is a 2‐bp deletion among three tandem GA repeats (296‐GAGAGAG‐302) within exon 2 resulting in a frameshift at codon 101 and the introduction of a termination codon at position 109 (often referred to as S109X). The actual 2 bp deleted within this sequence cannot be defined, consequently this mutation has been referred to as c.301_302delAG and c.296delGA in different reports Citation36, Citation37, Citation45, Citation57, Citation58, and probably represents a mutational hot spot within the gene, rather than a single common founder mutation Citation38. Furthermore, the c.301_302delAG and c.150delA mutations combined represent approximately 97% of all known mutations in PROP1. Reynaud et al. Citation59 reported the first PROP1 mutation downstream of the homeodomain involving a substitution of a tryptophan residue for a stop codon at position 194 (W194X) in the transactivation domain, with the resultant mutant protein showing only 34% activity compared to wild type PROP1. Subsequently two other mutations have been reported downstream of the homeodomain, including an individual with panhypopituitarism who was a compound heterozygote for c.150delA and a novel single base pair deletion, c.629delC Citation53, in addition to two sisters with GH, TSH, prolactin and gonadotropin deficiency with only slight pituitary hypoplasia who were homozygous for an insertion mutation in codon 156 (c.467insT) Citation52. Both these mutations result in frameshifts downstream of the homeodomain and are predicted to adversely affect the transactivation domain of the protein.
Table II. Reported mutations in the PROP1 gene.
Recessive inheritance of mutations in PROP1 is typically associated with GH, TSH, PRL and gonadotropin deficiencies, although the time of initiation and severity of pituitary hormone deficiencies is highly variable. Most patients present with early‐onset GH deficiency and growth retardation; however, normal growth in early childhood has been reported in a patient who attained a normal final height without GH replacement therapy Citation59, Citation60. In this case the patient presented with obesity and hypogonadotropic hypogonadism with the evolution of other hormonal deficiencies, including GH, later in life. TSH deficiency is also highly variable and has been reported as the first presenting symptom in some cases Citation37, Citation61, Citation62, while others show delayed TSH deficiency which may not be present at birth Citation43, Citation57, Citation61. Individuals with PROP1 mutations exhibit normal ACTH/cortisol levels in early life but often demonstrate an evolving cortisol deficiency that is strongly and significantly correlated with increasing age Citation54, Citation57, Citation63–65. However, patients as young as 6–7 years have also been described with cortisol deficiency Citation50, Citation54.
Although Prop1 is essential for the differentiation of gonadotropes in foetal life, the spectrum of gonadotropin deficiency is again extremely variable in patients with PROP1 mutations. Clinical variability can range from hypogonadism with complete lack of pubertal development to reports of spontaneous, albeit often delayed, onset of puberty with subsequent development of gonadotropin deficiency requiring hormone replacement Citation37, Citation43, Citation61, Citation64. Variation in timing and severity of gonadotropin deficiency suggests that hypogonadism in patients with PROP1 mutations is acquired rather than congenital, consistent with a role for PROP1 in maintenance or differentiation of gonadotropes as opposed to initial determination Citation41. Transgenic mice with persistent over‐expression of Prop1 in gonadotropes and thyrotropes have decreased expression of gonadotropins and exhibit transient hypogonadism. These mice subsequently recover from their abnormalities and at 3 months show normal size and fertility, suggesting that correct repression of Prop1 is important for the development and differentiation of gonadotropes Citation66.
The pituitary morphology in patients with PROP1 mutations is also highly variable; most individual reports have documented a normal pituitary stalk and posterior lobe, with a small or normal size anterior pituitary gland on MRI. However, in some cases, an enlarged anterior pituitary gland has been reported Citation36, Citation43, Citation57, Citation67. Longitudinal analyses of anterior pituitary size over time have revealed that a significant number of patients demonstrated pituitary enlargement in early childhood with subsequent regression and involution; thus ensuing MRI in older patients usually demonstrates anterior pituitary hypoplasia Citation62, Citation65. The pituitary enlargement consists of a mass lesion interposed between the anterior and posterior lobes, possibly originating from the intermediate lobe Citation62. Turton et al. (2005) recently demonstrated that the mass can wax and wane in size prior to eventual involution (Figure ) Citation39. To date, the underlying mechanism for the mass remains unknown. There has been only one report of a biopsy of the ‘tumour’, and the histology was non‐specific with the presence of amorphous material with no signs of apoptosis, and no recognizable cell lines Citation68, Citation69. Consequent to the highly variable phenotype associated with PROP1 mutations, no genotype‐phenotype correlation has been identified; furthermore phenotypic differences have been reported in siblings with identical mutations Citation61. It is possible that some missense mutations may retain some residual activity in vitro, which may influence the phenotype; however, the observation that Prop1 null mice display a nearly identical pituitary phenotype to the Ames dwarf mouse, suggests that the residual activity of the Ames mutation (S83P) is insignificant in vivoCitation35. Evidence of phenotypic differences of Prop1 mutations bred on different mouse strains suggests that unidentified genetic modifiers may play a role in the phenotypic variability observed in human patients. Such modifiers may include members of the Wnt/β‐catenin signalling pathway as it has recently been demonstrated that Prop1 is capable of interacting directly with β‐catenin, which acts as a co‐regulator for transcriptional activity of Prop1 with roles in direct repression of Hesx1 and simultaneous activation of Pou1f1 required for the determination of somatotrope, thyrotrope and corticotrope cell lineages Citation70. The evolving nature of hormone insufficiencies in patients with PROP1 mutations suggests a progressive decline in the anterior pituitary axis, indicating a need for continual monitoring of patients for the development of hormone insufficiencies that may not be apparent at initial presentation.
POU1F1
POU1F1 (OMIM 173110; previously known as PIT1) is a pituitary‐specific transcription factor belonging to the POU homeodomain family of transcription factors (named after the genes PIT1, OCT1 and unc‐86) characterized by a highly conserved DNA binding domain consisting of a POU‐specific domain and a POU homeodomain. In the mouse, Pou1f1 is expressed relatively late during pituitary development (14.5 dpc), and expression persists throughout postnatal life and adulthood, restricted to the anterior pituitary lobe Citation71. Pou1f1 is essential for the development of somatotrope, lactotrope and thyrotrope cell lineages in the anterior pituitary Citation72, and for the subsequent expression of the GH‐1 (OMIM 139250), prolactin (PRL; OMIM 176760) and TSH‐β (OMIM 188540) genes between 15.5 and 17 dpc Citation73. Two naturally occurring murine models have shed light on the role of Pou1f1 in normal pituitary development. In the Snell dwarf (dw) mouse, a recessive point mutation (W261C) results in the absence of somatotropes, lactotropes and thyrotropes Citation74. A similar phenotype results in the Jackson dwarf mouse (dwJ) that harbours a recessive null mutation due to rearrangement of Pou1f1. Pou1f1 binding sites have also been found in the GHRHR (OMIM 139191) and the Pou1f1 gene itself Citation72, Citation75, and autoregulation is required to sustain gene expression once the Pou1f1 protein has reached a critical threshold Citation76.
The first mutation within POUIFI was identified by Tatsumi et al. (1992) in a child with GH, prolactin and profound TSH deficiency caused by homozygosity for a nonsense mutation within the gene Citation77. The majority of mutations identified in POU1F1 to date are recessive; however, in addition, a number of heterozygous point mutations have been reported Citation78. Of these the amino acid substitution R271W appears to be a ‘hot spot’ for POU1F1 mutations Citation78, and has been identified in several unrelated patients of different ethnic backgrounds Citation79–86. When co‐transfected with wild type POU1F1, this mutant protein prevented transcriptional activation by the wild type protein acting as a dominant negative Citation80, Citation87, although this has been disputed Citation88.
The spectrum of hormone deficiency can vary in patients with POU1F1 mutations; GH and prolactin deficiencies generally present early in life, however TSH deficiency can be highly variable with presentation later in childhood Citation89, Citation90. We have recently described a POU1F1 mutation in a 21‐year‐old with GH and prolactin deficiency who has normal thyroid function to date Citation91. Magnetic resonance imaging demonstrates a small or normal anterior pituitary with a normal posterior pituitary and infundibulum, and no midline abnormalities. Since the first report, a total of 27 POU1F1 mutations have been described including 22 recessive, and a complete gene deletion, and 5 dominant mutations in over 60 patients originating from 19 different countries (Table ), all of which have been associated with a broadly similar phenotype of GH, TSH and prolactin deficiency Citation92.
Table III. Mutations within the POU1F1 gene.
LHX3/LHX4
Lhx3 is a member of the LIM family of homeobox genes which are characterized by the presence of a unique cysteine/histidine‐rich zinc binding LIM domain. The protein contains two such tandemly repeated LIM motifs between the N terminus and the homeodomain, which are likely to be involved in protein‐protein interactions Citation93, Citation94. Lhx3 is one of the earliest transcription factors expressed within the developing pituitary, initially detectable with strong uniform expression in Rathke's pouch. Expression is maintained in the pouch and is subsequently restricted to fields fated to form the anterior and intermediate lobes. By 16.5 dpc, the gene is expressed in all regions of the developing anterior and intermediate pituitary, but not the posterior gland, and continued expression is essential for the establishment of hormone‐producing cell types. Expression persists throughout development and is also detected in the adult pituitary suggesting a role in maintenance of one or more of the mature anterior pituitary cell types Citation93, Citation95. In addition to the pituitary, Lhx3 is also detected transiently in regions of the developing spinal cord, prior to neural tube closure, and later within restricted regions of the hindbrain, in addition to cells in immediate proximity to the otic vesicles Citation95. Mice with a targeted homozygous disruption of Lhx3 die shortly after birth, although the cause of death is unknown, and exhibit pituitary aplasia, suggesting an essential role of Lhx3 in differentiation and proliferation of anterior pituitary cell lineages. Although Rathke's pouch is initially formed in Lhx3 null mice, a failure of proliferation and growth results in a lack of the anterior and intermediate lobes of the pituitary with depletion of all hormone‐producing cell types except corticotropes, which differentiate and express pro‐opiomelanocortin (POMC), but fail to proliferate Citation96, Citation97. The posterior pituitary appeared grossly unaffected, as did the hindbrain and spinal cord, and heterozygous mice were apparently normal and fertile Citation96.
Homozygous mutations in LHX3 (OMIM 600577) have currently been identified in five patients from three unrelated consanguineous families, all of which result in loss of LHX3 function Citation98–101. The patients presented with an endocrine phenotype similar to that observed in individuals with PROP1 mutations with a deficit in all anterior pituitary hormones except ACTH. This was additionally associated in all patients with a short, rigid, cervical spine with limited head rotation and trunk movement. As with PROP1‐deficient patients, pituitary morphology is variable between patients with LHX3 mutations, with two patients from one family exhibiting small anterior pituitaries with a normal posterior pituitary and midline structures on MRI. However, an additional individual from an unrelated family demonstrated a markedly enlarged anterior pituitary that was not evident in a previous MR scan performed ten years previously Citation98. Additionally, Bhangoo et al. (2006) recently reported a further patient with a hypointense lesion in the anterior pituitary consistent with a microadenoma Citation100.
Lhx4 is closely related to Lhx3 and is expressed in specific fields of the developing brain and spinal cord. Similar to Lhx3, Lhx4 is initially expressed throughout the invaginating Rathke's pouch; however, subsequent expression is transient and restricted to the future anterior lobe, whereas Lhx3 expression is maintained throughout the whole pouch. A similar pattern of transient expression of LHX4 (OMIM 602146) in the developing pituitary and spinal cord, with continuous expression of LHX3, has also been observed in human development Citation101. Null mutations of Lhx4 do not prevent pouch formation; however, the pouch is defective with reduced numbers of the various anterior pituitary cell types. Lhx3−/−, Lhx4−/− double mutant mice show a more severe phenotype than either single mutant with an early arrest of pituitary development; additionally a single normal copy of either Lhx3 or Lhx4 in murine development is sufficient for formation of a definitive pouch, thereby suggesting that these two genes may act in a redundant manner during early pituitary development Citation97.
To date only a single mutation within LHX4 has been reported in a three‐generation family, in which the mutation, which abolishes normal splicing, segregates in a dominant and fully penetrant manner. The probands presented with short stature and were found to be GH, TSH and ACTH deficient Citation102. Anterior pituitary hypoplasia was revealed by MRI with an ectopic posterior pituitary and absent pituitary stalk. However, other affected family members presented with short stature associated with isolated GH deficiency and a normal posterior pituitary. Additional manifestations included a poorly formed sella turcica and pointed cerebellar tonsils suggesting that LHX4 may be a factor that tightly co‐ordinates brain development and skull shaping. Machinis and Amselem (2005) demonstrated that LHX4 binds to a specific element in the promoter of the POU1F1 gene in vitro, and is capable of activating a reporter construct containing a fragment of the POU1F1 upstream regulatory sequence encompassing the LHX4 binding site. Mutant LHX4 proteins generated from the defect identified in the patients failed to bind to and subsequently activate the POU1F1 regulatory sequence, and did not interfere with the ability of the normal protein to activate transcription. This suggests that haploinsufficiency of LHX4 results in defective regulation of POU1F1 and subsequently the downstream activation of GH expression, for which POU1F1 is required, providing a mechanism at the molecular level for the GH deficiency and short stature in patients carrying the LHX4 mutation. However, the ACTH deficiency observed in these patients cannot be explained by a defect in POU1F1 expression suggesting that other LHX4‐dependent pathways exist independent of POU1F1 in the developing pituitary Citation103.
X‐linked hypopituitarism and SOX3
A number of pedigrees have been described with X‐linked hypopituitarism involving duplications of Xq26‐q27 Citation104–107, with the smallest described to date being approximately 690 Kb Citation108. The phenotype of affected male patients associated with Xq27 duplications comprises variable mental retardation and hypopituitarism associated with anterior pituitary hypoplasia, infundibular hypoplasia and an ectopic posterior pituitary, with variable abnormalities of the corpus callosum (Figure ). All duplications associated with hypopituitarism encompass the SOX3 gene (OMIM 313430), a developmental transcription factor expressed in the developing infundibulum and hypothalamus Citation107. Further implication of SOX3 in hypopituitarism comes from the identification of affected patients with expansion of a polyalanine tract within the gene Citation108, Citation109. Woods et al. (2005) demonstrated that an expansion of the tract within SOX3 by seven alanine residues results in partial loss of function of the mutant protein, possibly due to impaired nuclear localization, associated with panhypopituitarism, anterior pituitary hypoplasia, a hypoplastic infundibulum and an ectopic/undescended posterior pituitary Citation108. The previous polyalanine expansion described by Laumonnier et al. (expanded by 11 alanine residues) was associated with IGHD rather than panhypopituitarism Citation109. Additionally, a proportion of Sox3 null mice exhibit pituitary and hypothalamic defects, in addition to craniofacial abnormalities, reduced size and fertility and defects of central nervous system (CNS) midline structures Citation110. Consistent with the location of SOX3 on the X chromosome, all patients described to date harbouring SOX3 mutations are male with female carriers appearing clinically unaffected. Male patients presenting with Xq27 duplications encompassing SOX3, together with loss‐of‐function polyalanine expansion mutations, and the phenotype of Sox3 null mice, suggest that Sox3/SOX3 dosage is critical for normal hypothalamic‐pituitary development. Hence, both over‐ and under‐dosage of SOX3 are associated with infundibular hypoplasia and variable hypopituitarism.
SOX2
SOX2 (OMIM 184429) is a member of the SRY‐related high mobility group (HMG) box (SOX) genes which, based on HMG box homology, is grouped into the B1 subfamily with SOX1 (OMIM 602148) and SOX3. In the mouse, initial expression of Sox2 is detected at 2.5 dpc at the morula stage, and then in the inner cell mass of the blastocyst at 3.5 dpc. Later expression of Sox2, following gastrulation, is restricted to the presumptive neuroectoderm, and by 9.5 dpc it is expressed throughout the brain, CNS, sensory placodes, branchial arches, gut endoderm and the oesophagus and trachea Citation111, Citation112. Homozygous loss of Sox2 results in peri‐implantation lethality, whereas Sox2 heterozygous mice appear relatively normal but show a reduction in size and male fertility Citation113. Given this observation, Kelberman et al. (2006) investigated the role of Sox2 in murine pituitary development, showing that a proportion of heterozygous animals manifested a variable hypopituitary phenotype, with hypoplasia and abnormal morphology of the anterior pituitary gland with concomitant reduction in levels of GH, LH, ACTH and TSH Citation114.
Heterozygous de novo mutations in SOX2 were previously reported in human patients associated with bilateral anophthalmia or severe microphthalmia with additional abnormalities reported, including developmental delay, learning difficulties, oesophageal atresia and genital abnormalities Citation112, Citation115–118. More recently, Kelberman et al. (2006) reported six patients harbouring de novo heterozygous mutations in SOX2 resulting in loss of function of the mutant protein, four of which were previously unreported (c.60insG, c.387delC, Y160X and c.479delA). Clinical evaluation revealed that in addition to anophthalmia/microphthalmia, SOX2 mutations were also associated with anterior pituitary hypoplasia and hypogonadotropic hypogonadism, which resulted in the absence of puberty in all six patients and genital abnormalities in males. All affected individuals exhibited learning difficulties with other variable manifestations including hippocampal abnormalities, defects of the corpus callosum, oesophageal atresia and sensorineural hearing loss Citation114.
TBX19
TBX19 (OMIM 604614; also referred to as TPIT) is a member of the T‐box family of transcription factors that contain a homologous DNA binding domain (the T‐box) first identified in the mouse Brachyury (T) gene. Tbx19 is not expressed within Rathke's pouch, but by 11.5 dpc expression appears within the developing anterior pituitary gland, as well as a region of the ventral diencephalon. Later expression in the developing and adult murine pituitary is detected in only two cell types, the POMC (OMIM 176830) expressing corticotropes in the anterior lobe and melanotropes forming the intermediate lobe, and is essential for cell specific expression of the POMC gene and terminal differentiation of the pituitary corticotrope lineage Citation119–121.
Consistent with the exclusive expression in pituitary POMC cells, mutations in TBX19 are associated at high frequency with neonatal isolated ACTH deficiency, but never in cases of juvenile‐onset deficiency. Twelve independent mutations have been identified including nonsense, missense, frameshift and splicing mutations, in addition to a large genomic deletion of 5.2 Kb encompassing exons 2 and 3 of the gene Citation119, Citation122–125. All of these mutations have been shown or are predicted to result in loss of TBX19 function Citation121, Citation123, and all patients appear to be homozygous or compound heterozygous for TBX19 mutations, with unaffected heterozygous parents, implying a recessive mode of inheritance. Vallette‐Kasic et al. (2005) have reported the largest series to date, and demonstrated TBX19 mutations in 17 out of 27 patients from 21 unrelated families, suggesting mutations in the gene are the principal molecular cause of congenital neonatal isolated ACTH deficiency. Moreover, three patients were identified to carry only one mutant TBX19 allele, although family history implied recessive inheritance suggesting that additional, as yet unidentified, mutations may be present in regulatory regions of the gene in some cases Citation123. Patients with TBX19 mutations present with a homogeneous clinical phenotype with an endocrine profile consisting of very low basal plasma ACTH and cortisol levels, with no significant ACTH response to corticotrophin releasing hormone; however, plasma cortisol levels may respond to repeated ACTH administration, with normal function of all other pituitary axes. Severe hypoglycaemia, associated with seizures in some cases, and prolonged cholestatic jaundice are classically associated with ACTH deficiency presenting in the neonatal period Citation119, Citation123. Furthermore, Vallette‐Kasic et al. noted that in their series about 25% of families with segregating TBX19 mutations (5 of 21) suffered a neonatal death, suggesting that isolated ACTH deficiency may be an underestimated cause of neonatal death. Tbx19 null mice constitute a model of isolated ACTH deficiency similar to the phenotype observed in human patients with TBX19 mutations Citation122.
PITX2
Pitx2 is a paired‐like homeobox gene that is expressed in the foetal pituitary and in most cells of the adult gland. In the mouse, it plays an important role in development of Rathke's pouch and anterior pituitary gland. Pitx2 is first expressed in the mouse embryo in oral epithelium and oral ectoderm, with later expression in the developing Rathke's pouch in addition to the mesenchyme near the optic eminence, the basal plate of the CNS, the forelimbs and domains of the abdominal cavity. It appears to be required for pituitary development shortly after formation of the committed pouch Citation126. Additionally, Pitx2 is implicated in left‐right asymmetry. Mutations in PITX2 (OMIM 601542 also known as RIEG1) are associated with Rieger syndrome in humans Citation127, an autosomal dominant condition with variable manifestations including anomalies of the anterior chamber of the eye, dental hypoplasia, a protuberant umbilicus, mental retardation and pituitary abnormalities. Although hypopituitarism with a hypoplastic gland has been described in mice with a targeted mutation in Pitx2Citation128, it remains unclear if humans with PITX2 mutations have pituitary hormone deficiencies.
Midline defects and GLI2
Recently, the Sonic Hedgehog (SHH) signalling pathway has been implicated in more complex disorders of pituitary development. Mutations within SHH (OMIM 600725) are associated with holoprosencephaly Citation129. Three members of the Gli gene family of transcription factors have been implicated in the mediation of SHH signals, and heterozygous loss of function mutations within the GLI2 gene (OMIM 165230) have been identified in patients with holoprosencephaly Citation130. Phenotypic penetrance was variable, with the disorder transmitted through a parent carrying the mutation but showing no obvious phenotype in one family. In all affected individuals with GLI2 mutations, pituitary gland function was abnormal, accompanied by variable craniofacial abnormalities. Other features included postaxial polydactyly, single nares, single central incisor and partial agenesis of the corpus callosum.
Conclusions
Hypopituitarism is defined as a deficiency of one or several pituitary hormones and can vary in severity and age at presentation. Additionally, the hormone abnormalities may evolve with time necessitating frequent evaluation. These hormonal deficits can also be present as part of a syndrome, with patients manifesting extra‐pituitary abnormalities. Over the past decade, there has been an explosion in the knowledge of the genetic cascade that orchestrates hypothalamo‐pituitary development. Several transcription factors and signalling molecules are critical for cell differentiation and proliferation at a very early stage of gestation. The mouse has served as an excellent model for understanding the genetic basis of congenital hypopituitarism in humans, although the correlation between mouse and human disease phenotypes is variable. This candidate gene approach, based on mouse studies, has led to the identification of several human mutations that disrupt hypothalamo‐pituitary development resulting in specific patterns of hormone dysfunction.
Establishing the genotype can aid in the management of individual patients with hypopituitarism. For example, identification of a mutation in PROP1 in a patient with an enlarged anterior pituitary may prevent the patient undergoing further invasive procedures, since it is now well established that the enlarged pituitary will undergo spontaneous involution. Additionally, identification of a mutation within POU1F1 predicts that cortisol and gonadotropin secretion will remain normal in the patient. Identification of the genotype can also aid in genetic counselling and early diagnosis, particularly in autosomal dominant POU1F1 mutations.
However, no genetic aetiology has been established to date in most patients with hypopituitarism. Genetic screening of several large cohorts of patients with CPHD has revealed that only a small percentage of individuals harbour mutations in the genes encoding transcription factors reviewed here Citation39, Citation53, Citation92, Citation131, Citation132, and given that a number of these patients may represent familial cases, it is clear that many genes implicated in hypopituitarism remain to be identified. Ongoing studies into the identification of such additional genes and molecules involved in pituitary development, for example the transcription factors IkarosCitation133 and Foxl2Citation134 and components of the Wnt/β‐catenin signalling pathway Citation70, Citation135, which have recently been shown to have roles in pituitary development in the mouse, and characterization of these, will in future enable a greater understanding of hypopituitarism.
References
- Takuma N., Sheng H. Z., Furuta Y., Ward J. M., Sharma K., Hogan L. M., et al. Formation of Rathke's pouch requires dual induction from the diencephalon. Development 1998; 125: 4835–40
- Dasen J. S., Rosenfeld M. G. Signaling mechanisms in pituitary morphogenesis and cell fate determination. Curr Opin Cell Biol 1999; 11: 669–77
- Dasen J. S., Rosenfeld M. G. Signaling and transcriptional mechanisms in pituitary development. Annu Rev Neurosci 2001; 24: 327–55
- Rizzoti K., Lovell‐Badge R. Early development of the pituitary gland: induction and shaping of Rathke's pouch. Rev Endocr Metab Disord 2005; 6: 161–72
- Cohen R. N. Update on genetic regulation of pituitary development. Pediatr Endocrinol Rev 2006; 3(Suppl 2)312–7
- GH Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab 2000; 85: 3990–3
- Parkin J. M. Incidence of growth hormone deficiency. Arch Dis Child 1974; 49: 904–5
- Vimpani G. V., Vimpani A. F., Lidgard G. P., Cameron E. H., Farquhar J. W. Prevalence of severe growth hormone deficiency. Br Med J 1977; 2: 427–30
- Lindsay R., Feldkamp M., Harris D., Robertson J., Rallison M. Utah Growth Study: growth standards and the prevalence of growth hormone deficiency. J Pediatr 1994; 125: 29–35
- Bao X. L., Shi Y. F., Du Y. C., Liu R., Deng J. Y., Gao S. M. Prevalence of growth hormone deficiency of children in Beijing. Chin Med J (Engl) 1992; 105: 401–5
- Thomas M., Massa G., Craen M., de Z. F., Bourguignon J. P., Heinrichs C., et al. Prevalence and demographic features of childhood growth hormone deficiency in Belgium during the period 1986–2001. Eur J Endocrinol 2004; 151: 67–72
- Phillips J. A III., Cogan J. D. Genetic basis of endocrine disease. 6. Molecular basis of familial human growth hormone deficiency. J Clin Endocrinol Metab 1994; 78: 11–6
- Patel L., McNally R. J., Harrison E., Lloyd I. C., Clayton P. E. Geographical distribution of optic nerve hypoplasia and septo‐optic dysplasia in Northwest England. J Pediatr 2006; 148: 85–8
- Tornqvist K., Ericsson A., Kallen B. Optic nerve hypoplasia: Risk factors and epidemiology. Acta Ophthalmol Scand 2002; 80: 300–4
- Hermesz E., Mackem S., Mahon K. A. Rpx: a novel anterior‐restricted homeobox gene progressively activated in the prechordal plate, anterior neural plate and Rathke's pouch of the mouse embryo. Development 1996; 122: 41–52
- Thomas P. Q., Johnson B. V., Rathjen J., Rathjen P. D. Sequence, genomic organization, and expression of the novel homeobox gene Hesx1. J Biol Chem 1995; 270: 3869–75
- Dasen J. S., Barbera J. P., Herman T. S., Connell S. O., Olson L., Ju B., et al. Temporal regulation of a paired‐like homeodomain repressor/TLE corepressor complex and a related activator is required for pituitary organogenesis. Genes Dev 2001; 15: 3193–207
- Sornson M. W., Wu W., Dasen J. S., Flynn S. E., Norman D. J., O'Connell S. M., et al. Pituitary lineage determination by the Prophet of Pit‐1 homeodomain factor defective in Ames dwarfism. Nature 1996; 384: 327–33
- Thomas P., Beddington R. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol 1996; 6: 1487–96
- Chou S. J., Hermesz E., Hatta T., Feltner D., El‐Hodiri H. M., Jamrich M., et al. Conserved regulatory elements establish the dynamic expression of Rpx/HesxI in early vertebrate development. Dev Biol 2006; 292: 533–45
- Dattani M. T., Martinez‐Barbera J. P., Thomas P. Q., Brickman J. M., Gupta R., Martensson I. L., et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo‐optic dysplasia in human and mouse. Nat Genet 1998; 19: 125–33
- Brickman J. M., Clements M., Tyrell R., McNay D., Woods K., Warner J., et al. Molecular effects of novel mutations in Hesx1/HESX1 associated with human pituitary disorders. Development 2001; 128: 5189–99
- Wales J. K., Quarrell O. W. Evidence for possible Mendelian inheritance of septo‐optic dysplasia. Acta Paediatr 1996; 85: 391–2
- Carvalho L. R., Woods K. S., Mendonca B. B., Marcal N., Zamparini A. L., Stifani S., et al. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor‐corepressor interaction. J Clin Invest 2003; 112: 1192–201
- Sobrier M. L., Netchine I., Heinrichs C., Thibaud N., Vie‐Luton M. P., Van V. G., et al. Alu‐element insertion in the homeodomain of HESX1 and aplasia of the anterior pituitary. Hum Mutat 2005; 25: 503
- Sobrier M. L., Maghnie M., Vie‐Luton M. P., Secco A., di Iorgi N., Lorini R., et al. Novel HESX1 mutations associated with a life‐threatening neonatal phenotype, pituitary aplasia, but normally located posterior pituitary and no optic nerve abnormalities. J Clin Endocrinol Metab, 2006 Aug 29 [Epub ahead of print.]
- Arrigo T., Wasniewska M., De Luca F., Valenzise M., Lombardo F., Vivenza D., et al. Congenital adenohypophysis aplasia: clinical features and analysis of the transcriptional factors for embryonic pituitary development. J Endocrinol Invest 2006; 29: 208–13
- Thomas P. Q., Dattani M. T., Brickman J. M., McNay D., Warne G., Zacharin M., et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo‐optic dysplasia. Hum Mol Genet 2001; 10: 39–45
- Cohen R. N., Cohen L. E., Botero D., Yu C., Sagar A., Jurkiewicz M., et al. Enhanced repression by HESX1 as a cause of hypopituitarism and septooptic dysplasia. J Clin Endocrinol Metab 2003; 88: 4832–9
- Tajima T., Hattorri T., Nakajima T., Okuhara K., Sato K., Abe S., et al. Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J Clin Endocrinol Metab 2003; 88: 45–50
- Gage P. J., Brinkmeier M. L., Scarlett L. M., Knapp L. T., Camper S. A., Mahon K. A. The Ames dwarf gene, df, is required early in pituitary ontogeny for the extinction of Rpx transcription and initiation of lineage‐specific cell proliferation. Mol Endocrinol 1996; 10: 1570–81
- Ward R. D., Raetzman L. T., Suh H., Stone B. M., Nasonkin I. O., Camper S. A. Role of PROP1 in pituitary gland growth. Mol Endocrinol 2005; 19: 698–710
- Ward R. D., Stone B. M., Raetzman L. T., Camper S. A. Cell Proliferation and Vascularization in Mouse Models of Pituitary Hormone Deficiency. Mol Endocrinol 2006; 20: 1378–90
- Tang K., Bartke A., Gardiner C. S., Wagner T. E., Yun J. S. Gonadotropin secretion, synthesis, and gene expression in human growth hormone transgenic mice and in Ames dwarf mice. Endocrinology 1993; 132: 2518–24
- Nasonkin I. O., Ward R. D., Raetzman L. T., Seasholtz A. F., Saunders T. L., Gillespie P. J., et al. Pituitary hypoplasia and respiratory distress syndrome in Prop1 knockout mice. Hum Mol Genet 2004; 13: 2727–35
- Wu W., Cogan J. D., Pfaffle R. W., Dasen J. S., Frisch H., O'Connell S. M., et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet 1998; 18: 147–9
- Deladoey J., Fluck C., Buyukgebiz A., Kuhlmann B. V., Eble A., Hindmarsh P. C., et al. ‘Hot spot’ in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 1999; 84: 1645–50
- Cogan J. D., Wu W., Phillips J. A., Arnhold I. J., Agapito A., Fofanova O. V., et al. The PROP1 2‐base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998; 83: 3346–9
- Turton J. P. G., Mehta A., Raza J., Woods K. S., Tiulpakov A., Cassar J., et al. Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD). Clin Endocrinol (Oxf) 2005; 63: 10–8
- Duquesnoy P., Roy A., Dastot F., Ghali I., Teinturier C., Netchine I., et al. Human Prop‐1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency. FEBS Lett 1998; 437: 216–20
- Parks J. S., Brown M. R., Hurley D. L., Phelps C. J., Wajnrajch M. P. Heritable disorders of pituitary development. J Clin Endocrinol Metab 1999; 84: 4362–70
- Voutetakis A., Maniati‐Christidi M., Kanaka‐Gantenbein C., Dracopoulou M., Argyropoulou M., Livadas S., et al. Prolonged jaundice and hypothyroidism as the presenting symptoms in a neonate with a novel Prop1 gene mutation (Q83X). Eur J Endocrinol 2004; 150: 257–64
- Vallette‐Kasic S., Barlier A., Teinturier C., Diaz A., Manavela M., Berthezene F., et al. PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab 2001; 86: 4529–35
- Osorio M. G., Kopp P., Marui S., Latronico A. C., Mendonca B. B., Arnhold I. J. Combined pituitary hormone deficiency caused by a novel mutation of a highly conserved residue (F88S) in the homeodomain of PROP‐1. J Clin Endocrinol Metab 2000; 85: 2779–85
- Lebl J., Vosahlo J., Pfaeffle R. W., Stobbe H., Cerna J., Novotna D., et al. Auxological and endocrine phenotype in a population‐based cohort of patients with PROP1 gene defects. Eur J Endocrinol 2005; 153: 389–96
- Vieira T. C., Dias da Silva M. R., Cerutti J. M., Brunner E., Borges M., Arnaldi L. T., et al. Familial combined pituitary hormone deficiency due to a novel mutation R99Q in the hot spot region of Prophet of Pit‐1 presenting as constitutional growth delay. J Clin Endocrinol Metab 2003; 88: 38–44
- Paracchini R., Giordano M., Corrias A., Mellone S., Matarazzo P., Bellone J., et al. Two new PROP1 gene mutations responsible for compound pituitary hormone deficiency. Clin Genet 2003; 64: 142–7
- Lemos M. C., Gomes L., Bastos M., Leite V., Limbert E., Carvalho D., et al. PROP1 gene analysis in Portuguese patients with combined pituitary hormone deficiency. Clin Endocrinol (Oxf) 2006; 65: 479–85
- Fofanova O. V., Takamura N., Kinoshita E., Parks J. S., Brown M. R., Peterkova V. A., et al. A mutational hot spot in the Prop‐1 gene in Russian children with combined pituitary hormone deficiency. Pituitary 1998; 1: 45–9
- Agarwal G., Bhatia V., Cook S., Thomas P. Q. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J Clin Endocrinol Metab 2000; 85: 4556–61
- Tatsumi K. I., Kikuchi K., Tsumura K., Amino N. A novel PROP1 gene mutation (157delA) in Japanese siblings with combined anterior pituitary hormone deficiency. Clin Endocrinol (Oxf) 2004; 61: 635–40
- Nose O., Tatsumi K., Nakano Y., Amino N. Congenital combined pituitary hormone deficiency attributable to a novel PROP1 mutation (467insT). J Pediatr Endocrinol Metab 2006; 19: 491–8
- Reynaud R., Gueydan M., Saveanu A., Vallette‐Kasic S., Enjalbert A., Brue T., et al. Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J Clin Endocrinol Metab 2006; 91: 3329–36
- Bottner A., Keller E., Kratzsch J., Stobbe H., Weigel J. F., Keller A., et al. PROP1 Mutations Cause Progressive Deterioration of Anterior Pituitary Function including Adrenal Insufficiency: A Longitudinal Analysis. J Clin Endocrinol Metab 2004; 89: 5256–65
- Abrao M. G., Leite M. V., Carvalho L. R., Billerbeck A. E., Nishi M. Y., Barbosa A. S., et al. Combined pituitary hormone deficiency (CPHD) due to a complete PROP1 deletion. Clin Endocrinol (Oxf) 2006; 65: 294–300
- Reynaud R., Chadli‐Chaieb M., Vallette‐Kasic S., Barlier A., Sarles J., Pellegrini‐Bouiller I., et al. A Familial Form of Congenital Hypopituitarism Due to a PROP1 Mutation in a Large Kindred: Phenotypic and in Vitro Functional Studies. J Clin Endocrinol Metab 2004; 89: 5779–86
- Mendonca B. B., Osorio M. G., Latronico A. C., Estefan V., Lo L. S., Arnhold I. J. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301,G302 in the PROP1 gene. J Clin Endocrinol Metab 1999; 84: 942–5
- Fofanova O., Takamura N., Kinoshita E., Parks J. S., Brown M. R., Peterkova V. A., et al. Compound heterozygous deletion of the PROP‐1 gene in children with combined pituitary hormone deficiency. J Clin Endocrinol Metab 1998; 83: 2601–4
- Reynaud R., Barlier A., Vallette‐Kasic S., Saveanu A., Guillet M. P., Simonin G., et al. An uncommon phenotype with familial central hypogonadism caused by a novel PROP1 gene mutant truncated in the transactivation domain. J Clin Endocrinol Metab 2005; 90: 4880–7
- Arroyo A., Pernasetti F., Vasilyev V. V., Amato P., Yen S. S., Mellon P. L. A unique case of combined pituitary hormone deficiency caused by a PROP1 gene mutation (R120C) associated with normal height and absent puberty. Clin Endocrinol (Oxf) 2002; 57: 283–91
- Fluck C., Deladoey J., Rutishauser K., Eble A., Marti U., Wu W., et al. Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting in the substitution of Arg–>Cys at codon 120 (R120C). J Clin Endocrinol Metab 1998; 83: 3727–34
- Voutetakis A., Argyropoulou M., Sertedaki A., Livadas S., Xekouki P., Maniati‐Christidi M., et al. Pituitary magnetic resonance imaging in 15 patients with Prop1 gene mutations: pituitary enlargement may originate from the intermediate lobe. J Clin Endocrinol Metab 2004; 89: 2200–6
- Asteria C., Oliveira J. H., Abucham J., Beck‐Peccoz P. Central hypocortisolism as part of combined pituitary hormone deficiency due to mutations of PROP‐1 gene. Eur J Endocrinol 2000; 143: 347–52
- Pernasetti F., Toledo S. P., Vasilyev V. V., Hayashida C. Y., Cogan J. D., Ferrari C., et al. Impaired adrenocorticotropin‐adrenal axis in combined pituitary hormone deficiency caused by a two‐base pair deletion (301‐302delAG) in the prophet of Pit‐1 gene. J Clin Endocrinol Metab 2000; 85: 390–7
- Riepe F. G., Partsch C. J., Blankenstein O., Monig H., Pfaffle R. W., Sippell W. G. Longitudinal imaging reveals pituitary enlargement preceding hypoplasia in two brothers with combined pituitary hormone deficiency attributable to PROP1 mutation. J Clin Endocrinol Metab 2001; 86: 4353–7
- Vesper A. H., Raetzman L. T., Camper S. A. Role of prophet of Pit1 (PROP1) in gonadotrope differentiation and puberty. Endocrinology 2006; 147: 1654–63
- Fofanova O., Takamura N., Kinoshita E., Vorontsov A., Vladimirova V., Dedov I., et al. MR imaging of the pituitary gland in children and young adults with congenital combined pituitary hormone deficiency associated with PROP1 mutations. AJR Am J Roentgenol 2000; 174: 555–9
- Parks J. S., Brown M. R., Baumbach L., Sanchez J. C., Stanley C. A., Gianella‐Neto D., et al. Natural history and molecular mechanisms of hypopituitarism with large sella turcica. Presented at the American Endocrine Society Meeting, New Orleans, June 24–27, 1998. Abstract no. P3‐409. 1998; 470
- Cushman L. J., Watkins‐Chow D. E., Brinkmeier M. L., Raetzman L. T., Radak A. L., Lloyd R. V., et al. Persistent Prop1 expression delays gonadotrope differentiation and enhances pituitary tumor susceptibility. Hum Mol Genet 2001; 10: 1141–53
- Olson L. E., Tollkuhn J., Scafoglio C., Krones A., Zhang J., Ohgi K. A., et al. Homeodomain‐mediated beta‐catenin‐dependent switching events dictate cell‐lineage determination. Cell 2006; 125: 593–605
- Bodner M., Castrillo J. L., Theill L. E., Deerinck T., Ellisman M., Karin M. The pituitary‐specific transcription factor GHF‐1 is a homeobox‐containing protein. Cell 1988; 55: 505–18
- Andersen B., Rosenfeld M. G. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev 2001; 22: 2–35
- Rhodes S. J., DiMattia G. E., Rosenfeld M. G. Transcriptional mechanisms in anterior pituitary cell differentiation. Curr Opin Genet Dev 1994; 4: 709–17
- Li S., Crenshaw E. B III., Rawson E. J., Simmons D. M., Swanson L. W., Rosenfeld M. G. Dwarf locus mutants lacking three pituitary cell types result from mutations in the POU‐domain gene pit‐1. Nature 1990; 347: 528–33
- Andersen B., Rosenfeld M. G. Pit‐1 determines cell types during development of the anterior pituitary gland. A model for transcriptional regulation of cell phenotypes in mammalian organogenesis. J Biol Chem 1994; 269: 29335–8
- Rhodes S. J., Chen R., DiMattia G. E., Scully K. M., Kalla K. A., Lin S. C., et al. A tissue‐specific enhancer confers Pit‐1‐dependent morphogen inducibility and autoregulation on the pit‐1 gene. Genes Dev 1993; 7: 913–32
- Tatsumi K., Miyai K., Notomi T., Kaibe K., Amino N., Mizuno Y., et al. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet 1992; 1: 56–8
- Cohen L. E., Wondisford F. E., Salvatoni A., Maghnie M., Brucker‐Davis F., Weintraub B. D., et al. A ‘hot spot’ in the Pit‐1 gene responsible for combined pituitary hormone deficiency: clinical and molecular correlates. J Clin Endocrinol Metab 1995; 80: 679–84
- Ohta K., Nobukuni Y., Mitsubuchi H., Fujimoto S., Matsuo N., Inagaki H., et al. Mutations in the Pit‐1 gene in children with combined pituitary hormone deficiency. Biochem Biophys Res Commun 1992; 189: 851–5
- Radovick S., Nations M., Du Y., Berg L. A., Weintraub B. D., Wondisford F. E. A mutation in the POU‐homeodomain of Pit‐1 responsible for combined pituitary hormone deficiency. Science 1992; 257: 1115–8
- Okamoto N., Wada Y., Ida S., Koga R., Ozono K., Chiyo H., et al. Monoallelic expression of normal mRNA in the PIT1 mutation heterozygotes with normal phenotype and biallelic expression in the abnormal phenotype. Hum Mol Genet 1994; 3: 1565–8
- de Zegher F., Pernasetti F., Vanhole C., Devlieger H., Van den Berghe G., Martial J. A. The prenatal role of thyroid hormone evidenced by fetomaternal Pit‐1 deficiency. J Clin Endocrinol Metab 1995; 80: 3127–30
- Holl R. W., Pfaffle R., Kim C., Sorgo W., Teller W. M., Heimann G. Combined pituitary deficiencies of growth hormone, thyroid stimulating hormone and prolactin due to Pit‐1 gene mutation: a case report. Eur J Pediatr 1997; 156: 835–7
- Aarskog D., Eiken H. G., Bjerknes R., Myking O. L. Pituitary dwarfism in the R271W Pit‐1 gene mutation. Eur J Pediatr 1997; 156: 829–34
- Rodrigues Martineli A. M., Braga M., De Lacerda L., Raskin S., Graf H. Description of a Brazilian patient bearing the R271W Pit‐1 gene mutation. Thyroid 1998; 8: 299–304
- Ward L., Chavez M., Huot C., Lecocq P., Collu R., Decarie J. C., et al. Severe congenital hypopituitarism with low prolactin levels and age‐dependent anterior pituitary hypoplasia: a clue to a PIT‐1 mutation. J Pediatr 1998; 132: 1036–8
- Cohen R. N., Brue T., Naik K., Houlihan C. A., Wondisford F. E., Radovick S. The role of CBP/p300 interactions and Pit‐1 dimerization in the pathophysiological mechanism of combined pituitary hormone deficiency. J Clin Endocrinol Metab 2006; 91: 239–47
- Kishimoto M., Okimura Y., Fumoto M., Iguchi G., Iida K., Kaji H., et al. The R271W mutant form of Pit‐1 does not act as a dominant inhibitor of Pit‐1 action to activate the promoters of GH and prolactin genes. Eur J Endocrinol 2003; 148: 619–25
- Pfaffle R. W., DiMattia G. E., Parks J. S., Brown M. R., Wit J. M., Jansen M., et al. Mutation of the POU‐specific domain of Pit‐1 and hypopituitarism without pituitary hypoplasia. Science 1992; 257: 1118–21
- Pfaffle R. W., Martinez R., Kim C., Frisch H., Lebl J., Otten B., et al. GH and TSH deficiency. Exp Clin Endocrinol Diabetes 1997; 105(Suppl 4)1–5
- Turton J. P. G., Reynaud R., Mehta A., Torpiano J., Saveanu A., Woods K. S., et al. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. J Clin Endocrinol Metab 2005; 90: 4762–70
- Cohen L. E., Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev 2002; 23: 431–42
- Bach I., Rhodes S. J., Pearse R. V., Heinzel T., Gloss B., Scully K. M., et al. P‐Lim, a LIM homeodomain factor, is expressed during pituitary organ and cell commitment and synergizes with Pit‐1. Proc Natl Acad Sci U S A 1995; 92: 2720–4
- Schmitt S., Biason‐Lauber A., Betts D., Schoenle E. J. Genomic structure, chromosomal localization, and expression pattern of the human LIM‐homeobox3 (LHX 3) gene. Biochem Biophys Res Commun 2000; 274: 49–56
- Zhadanov A. B., Bertuzzi S., Taira M., Dawid I. B., Westphal H. Expression pattern of the murine LIM class homeobox gene Lhx3 in subsets of neural and neuroendocrine tissues. Dev Dyn 1995; 202: 354–64
- Sheng H. Z., Zhadanov A. B., Mosinger B., Fujii T., Bertuzzi S., Grinberg A., et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science 1996; 272: 1004–7
- Sheng H. Z., Moriyama K., Yamashita T., Li H., Potter S. S., Mahon K. A., et al. Multistep control of pituitary organogenesis. Science 1997; 278: 1809–12
- Netchine I., Sobrier M. L., Krude H., Schnabel D., Maghnie M., Marcos E., et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet 2000; 25: 182–6
- Sloop K. W., Parker G. E., Hanna K. R., Wright H. A., Rhodes S. J. LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene 2001; 265: 61–9
- Bhangoo A. P., Hunter C. S., Savage J. J., Anhalt H., Pavlakis S., Walvoord E. C., et al. Clinical case seminar: A Novel LHX3 Mutation Presenting as Combined Pituitary Hormonal Deficiency. J Clin Endocrinol Metab 2006; 91: 747–53
- Sobrier M. L., Attie‐Bitach T., Netchine I., Encha‐Razavi F., Vekemans M., Amselem S. Pathophysiology of syndromic combined pituitary hormone deficiency due to a LHX3 defect in light of LHX3 and LHX4 expression during early human development. Gene Expr Patterns 2004; 5: 279–84
- Machinis K., Pantel J., Netchine I., Leger J., Camand O. J. A., Sobrier M. L., et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet 2001; 69: 961–8
- Machinis K., Amselem S. Functional relationship between LHX4 and POU1F1 in light of the LHX4 mutation identified in patients with pituitary defects. J Clin Endocrinol Metab 2005; 90: 5456–62
- Hamel B. C., Smits A. P., Otten B. J., van den Helm B., Ropers H. H., Mariman E. C. Familial X‐linked mental retardation and isolated growth hormone deficiency: clinical and molecular findings. Am J Med Genet 1996; 64: 35–41
- Hol F. A., Schepens M. T., van Beersum S. E., Redolfi E., Affer M., Vezzoni P., et al. Identification and characterization of an Xq26‐q27 duplication in a family with spina bifida and panhypopituitarism suggests the involvement of two distinct genes. Genomics 2000; 69: 174–81
- Solomon N. M., Nouri S., Warne G. L., Lagerstrom‐Fermer M., Forrest S. M., Thomas P. Q. Increased gene dosage at Xq26‐q27 is associated with X‐linked hypopituitarism. Genomics 2002; 79: 553–9
- Solomon N. M., Ross S. A., Morgan T., Belsky J. L., Hol F. A., Karnes P. S., et al. Array comparative genomic hybridisation analysis of boys with X linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet 2004; 41: 669–78
- Woods K. S., Cundall M., Turton J., Rizotti K., Mehta A., Palmer R., et al. Over‐ and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet 2005; 76: 833–49
- Laumonnier F., Ronce N., Hamel B. C. J., Thomas P., Lespinasse J., Raynaud M., et al. Transcription factor SOX3 is involved in X‐linked mental retardation with growth hormone deficiency. Am J Hum Genet 2002; 71: 1450–5
- Rizzoti K., Brunelli S., Carmignac D., Thomas P. Q., Robinson I. C., Lovell‐Badge R. SOX3 is required during the formation of the hypothalamo‐pituitary axis. Nat Genet 2004; 36: 247–55
- Wood H. B., Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre‐gastrulation to early somite stages. Mech Dev 1999; 86: 197–201
- Williamson K. A., Hever A. M., Rainger J., Rogers R. C., Magee A., Fiedler Z., et al. Mutations in SOX2 Cause Anophthalmia‐Esophageal‐Genital (AEG) Syndrome. Hum Mol Genet 2006; 15: 1413–22
- Avilion A. A., Nicolis S. K., Pevny L. H., Perez L., Vivian N., Lovell‐Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 2003; 17: 126–40
- Kelberman D., Rizzoti K., Avilion A., Bitner‐Glindzicz M., Cianfarani S., Collins J., et al. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo‐pituitary‐gonadal axis in mice and humans. J Clin Invest 2006; 116: 2442–55
- Fantes J., Ragge N. K., Lynch S. A., Mcgill N. I., Collin J. R. O., Howard‐Peebles P. N., et al. Mutations in SOX2 cause anophthalmia. Nat Genet 2003; 33: 461–3
- Ragge N. K., Lorenz B., Schneider A., Bushby K., de Sanctis L., de Sanctis U., et al. SOX2 anophthalmia syndrome. Am J Med Genet A 2005; 135A: 1–7
- Hagstrom S. A., Pauer G. J. T., Reid J., Simpson E., Crowe S., Maumenee I. H., et al. SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. Am J Med Genet A 2005; 138A: 95–8
- Zenteno J. C., Gascon‐Guzman G., Tovilla‐Canales J. L. Bilateral anophthalmia and brain malformations caused by a 20‐bp deletion in the SOX2 gene. Clin Genet 2005; 68: 564–6
- Lamolet B., Pulichino A., Lamonerie T., Gauthier Y., Brue T., Enjalbert A., et al. A Pituitary Cell‐Restricted T Box Factor, Tpit, Activates POMC Transcription in Cooperation with Pitx Homeoproteins. Cell 2001; 104: 849–59
- Liu J., Lin C., Gleiberman A., Ohgi K. A., Herman T., Huang H. P., et al. Tbx19, a tissue‐selective regulator of POMC gene expression. Proc Natl Acad Sci U S A 2001; 98: 8674–9
- Pulichino A. M., Vallette‐Kasic S., Tsai J. P., Couture C., Gauthier Y., Drouin J. Tpit determines alternate fates during pituitary cell differentiation. Genes Dev 2003; 17: 738–47
- Pulichino A. M., Vallette‐Kasic S., Couture C., Gauthier Y., Brue T., David M., et al. Human and mouse TPIT gene mutations cause early onset pituitary ACTH deficiency. Genes Dev 2003; 17: 711–6
- Vallette‐Kasic S., Brue T., Pulichino A. M., Gueydan M., Barlier A., David M., et al. Congenital isolated adrenocorticotropin deficiency: an underestimated cause of neonatal death, explained by TPIT gene mutations. J Clin Endocrinol Metab 2005; 90: 1323–31
- Metherell L. A., Savage M. O., Dattani M., Walker J., Clayton P. E., Farooqi I. S., et al. TPIT mutations are associated with early‐onset, but not late‐onset isolated ACTH deficiency. Eur J Endocrinol 2004; 151: 463–5
- Weintrob N., Drouin J., Vallette‐Kasic S., Taub E., Marom D., Lebenthal Y., et al. Low estriol levels in the maternal triple‐marker screen as a predictor of isolated adrenocorticotropic hormone deficiency caused by a new mutation in the TPIT gene. Pediatrics 2006; 117: e322–e327
- Suh H., Gage P. J., Drouin J., Camper S. A. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development 2002; 129: 329–37
- Semina E. V., Reiter R., Leysens N. J., Alward W. L., Small K. W., Datson N. A., et al. Cloning and characterization of a novel bicoid‐related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996; 14: 392–9
- Lin C. R., Kioussi C., O'Connell S., Briata P., Szeto D., Liu F., et al. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 1999; 401: 279–82
- Roessler E., Belloni E., Gaudenz K., Jay P., Berta P., Scherer S. W., et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 1996; 14: 357–60
- Roessler E., Du Y. Z., Mullor J. L., Casas E., Allen W. P., Gillessen‐Kaesbach G., et al. Loss‐of‐function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly‐like features. Proc Natl Acad Sci U S A 2003; 100: 13424–9
- Fofanova O. V., Takamura N., Kinoshita E., Yoshimoto M., Tsuji Y., Peterkova V. A., et al. Rarity of PIT1 involvement in children from Russia with combined pituitary hormone deficiency. Am J Med Genet 1998; 77: 360–5
- Rainbow L. A., Rees S. A., Shaikh M. G., Shaw N. J., Cole T., Barrett T. G., et al. Mutation analysis of POUF‐1, PROP‐1 and HESX‐1 show low frequency of mutations in children with sporadic forms of combined pituitary hormone deficiency and septo‐optic dysplasia. Clin Endocrinol (Oxf) 2005; 62: 163–8
- Ezzat S., Mader R., Fischer S., Yu S., Ackerley C., Asa S. L. An essential role for the hematopoietic transcription factor Ikaros in hypothalamic‐pituitary‐mediated somatic growth. Proc Natl Acad Sci U S A 2006; 103: 2214–9
- Ellsworth B. S., Egashira N., Haller J. L., Butts D. L., Cocquet J., Clay C. M., et al. FOXL2 in the Pituitary: Molecular, Genetic and Developmental Analysis. 0000, Mol Endocrinol. 2006 Jul 13; [Epub ahead of print]
- Douglas K. R., Brinkmeier M. L., Kennell J. A., Eswara P., Harrison T. A., Patrianakos A. I., et al. Identification of members of the Wnt signaling pathway in the embryonic pituitary gland. Mamm Genome 2001; 12: 843–51
- Salemi S., Besson A., Eble A., Gallati S., Pfaffle R. W., Mullis P. E. New N‐terminal located mutation (Q4ter) within the POU1F1‐gene (PIT‐1) causes recessive combined pituitary hormone deficiency and variable phenotype. Growth Horm IGF Res 2003; 13: 264–8
- Pellegrini‐Bouiller I., Belicar P., Barlier A., Gunz G., Charvet J. P., Jaquet P., et al. A new mutation of the gene encoding the transcription factor Pit‐1 is responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab 1996; 81: 2790–6
- McLennan K., Jeske Y., Cotterill A., Cowley D., Penfold J., Jones T., et al. Combined pituitary hormone deficiency in Australian children: clinical and genetic correlates. Clin Endocrinol (Oxf) 2003; 58: 785–94
- Hashimoto Y., Cisternino M., Cohen L. E. A novel nonsense mutation in the Pit‐1 gene: evidence for a gene dosage effect. J Clin Endocrinol Metab 2003; 88: 1241–7
- Wit J. M., Drayer N. M., Jansen M., Walenkamp M. J., Hackeng W. H., Thijssen J. H., et al. Total deficiency of growth hormone and prolactin, and partial deficiency of thyroid stimulating hormone in two Dutch families: a new variant of hereditary pituitary deficiency. Horm Res 1989; 32: 170–7
- Malvagia S., Poggi G. M., Pasquini E., Donati M. A., Pela I., Morrone A., et al. The de novo Q167K mutation in the POU1F1 gene leads to combined pituitary hormone deficiency in an Italian patient. Pediatr Res 2003; 54: 635–40
- Brown M. R., Parks J. S., Adess M. E., Rich B. H., Rosenthal I. M., Voss T. C., et al. Central hypothyroidism reveals compound heterozygous mutations in the Pit‐1 gene. Horm Res 1998; 49: 98–102
- Hendriks‐Stegeman B. I., Augustijn K. D., Bakker B., Holthuizen P., Der Vliet P. C., Jansen M. Combined pituitary hormone deficiency caused by compound heterozygosity for two novel mutations in the pou domain of the pit1/pou1f1 gene. J Clin Endocrinol Metab 2001; 86: 1545–50
- Gat‐Yablonski G., Lazar L., Pertzelan A., Phillip M. A novel mutation in PIT‐1: phenotypic variability in familial combined pituitary hormone deficiencies. J Pediatr Endocrinol Metab 2002; 15: 325–30
- Cohen L. E., Zanger K., Brue T., Wondisford F. E., Radovick S. Defective retinoic acid regulation of the Pit‐1 gene enhancer: A novel mechanism of combined pituitary hormone deficiency. Mol Endocrinol 1999; 13: 476–84
- Pernasetti F., Milner R. D., al Ashwal A. A., de Zegher F., Chavez V. M., Muller M., et al. Pro239Ser: a novel recessive mutation of the Pit‐1 gene in seven Middle Eastern children with growth hormone, prolactin, and thyrotropin deficiency. J Clin Endocrinol Metab 1998; 83: 2079–83
- Gat‐Yablonski G., Klar A., Hirsch D., Eliakim A., Lazar L., Hurvitz H., et al. Three novel mutations in POU1F1 in Israeli patients with combined pituitary hormone deficiency. J Pediatr Endocrinol Metab 2005; 18: 385–93
- Irie Y., Tatsumi K., Ogawa M., Kamijo T., Preeyasombat C., Suprasongsin C., et al. A novel E250X mutation of the PIT1 gene in a patient with combined pituitary hormone deficiency. Endocr J 1995; 42: 351–4
- Blankenstein O., Muhlenberg R., Kim C., Wuller S., Pfaffle R., Heimann G. A new C‐terminal located mutation (V272ter) in the PIT‐1 gene manifesting with severe congenital hypothyroidism. Possible functionality of the PIT‐1 C‐terminus. Horm Res 2001; 56: 81–6