Abstract
Background. Variants in the complement factor H gene (CFH) are associated with age‐related macular degeneration (AMD). CFH and five CFH‐related genes (CFHR1‐5) lie within the regulators of complement activation (RCA) locus on chromosome 1q32.
Aims and Methods. In this study, the structural and evolutionary relationships between these genes and AMD was refined using a combined genetic, molecular and immunohistochemical approach.
Results. We identify and characterize a large, common deletion that encompasses both the CFHR1 and CFHR3 genes. CFHR1, an abundant serum protein, is absent in subjects homozygous for the deletion. Genotyping analyses of AMD cases and controls from two cohorts demonstrates that deletion homozygotes comprise 1.1% of cases and 5.7% of the controls (chi‐square = 32.8; P = 1.6 E‐09). CFHR1 and CFHR3 transcripts are abundant in liver, but undetectable in the ocular retinal pigmented epithelium/choroid complex. AMD‐associated CFH/CFHR1/CFHR3 haplotypes are widespread in human populations.
Conclusion. The absence of CFHR1 and/or CFHR3 may account for the protective effects conferred by some CFH haplotypes. Moreover, the high frequencies of the 402H allele and the delCFHR1/CFHR3 alleles in African populations suggest an ancient origin for these alleles. The considerable diversity accumulated at this locus may be due to selection, which is consistent with an important role for the CFHR genes in innate immunity.
| Abbreviations | ||
| AMD | = | age‐related macular degeneration |
| CFH | = | complement factor H |
| CFHR | = | complement factor H‐related |
| RPE | = | retinal pigmented epithelium |
| SSCP | = | single strand conformation polymorphism |
| SDS‐PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| dNTP | = | deoxynucleotide triphosphate |
| PBS | = | phosphate buffered saline |
| HRP | = | horseradish peroxidase |
| TBST | = | Tris‐buffered saline Tween 20 |
| QPCR | = | quantitative polymerase chain reaction |
| MHC | = | major histocompatibility complex |
Introduction
As the leading cause of irreversible vision loss in the developed world, age‐related macular degeneration (AMD) affects an estimated 25% of individuals over the age of 75 Citation1, Citation2. A host of studies conducted over the past decade has provided compelling biological evidence that immune‐mediated processes and aberrant regulation of the complement alternative pathway contribute to the etiology of AMD Citation3–8. Definitive support for this new AMD paradigm has emerged from recent genetic studies that have shown a highly significant association between AMD and specific haplotypes of the complement factor H gene (CFH), which lies within the ‘regulators of complement activation’ (RCA) gene cluster on chromosome 1q25–q31 Citation9–12. Further support for the involvement of the complement cascade is provided by a related discovery that haplotypes of two additional complement components, factor B (CFB/FB) and complement component 2 (C2)—which lie within the major histocompatibility (MHC) class III region on 6p21—confer protection or increase the risk for AMD. Combined analyses of the CFH and CFB/C2 variants predict clinical outcome in approximately 74% of AMD cases Citation13.
Data related to the association of CFH with AMD in the Caucasian and Indian populations is consistent Citation14–27. Several recent reports, however, suggest that risk variants of the CFH gene in the Japanese and Chinese populations are either not associated with AMD, differ substantially from the Caucasian population, and/or occur at a substantially lower frequency Citation28–30. Although they are based upon relatively small cohorts, these reports indicate that additional genetic analyses are warranted across a wide variety of ethnic groups.
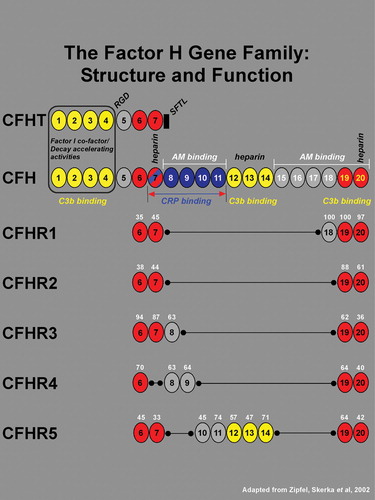
The CFH gene consists of 20 short consensus repeats (SCR), each of which encodes a functional domain of approximately 60 amino acids Citation31, Citation32. In addition to the full‐length transcript, there is an alternatively spliced, truncated gene product CFHT, which contain SCRs 1–7. Five additional members of the CFH gene family, represented by five separate genes, are designated as CFH‐related (CFHR) 1–5. Each of the five CFH‐related proteins contains a set of SCR domains, many of which are homologous to those in CFH (Figure ) Citation33. Most notably, the two most C‐terminal SCR domains are highly conserved within the CFH‐protein family. These domains contain a C3d/C3b binding site and a hot‐spot for mutations in atypical hemolytic uremic syndrome Citation34. All members of the factor H gene family appear to be synthesized primarily by cells in the liver and secreted into the circulation Citation32, Citation35.
Figure 1 TheCFH gene contains 20 short consensus repeats (SCR), or complement control modules, each of which encodes a functional domain of ∼60 amino acids. In addition to the full‐length transcript, there is an alternatively spliced, truncated gene product of CFH that has sequence identity with SCRs 1–7. An 8th exon spliced to SCR 7 in the truncated variant (black rectangle) encodes a stop codon, a unique untranslated region, and four C‐terminal amino acids (SFTL). The full length and truncated forms of CFH transcripts are referred to as variants 1 and 2, and the respective encoded proteins are designated as isoforms a and b. The 20 SCRs of CFH encode a number of functional domains. The complement factor I cofactor and decay‐accelerating activities reside within SCRs 1–4 (boxed). Three C3b binding sites are associated with SCRs 1–4, 12–14, and 19–20 (shown in yellow). A RGD sequence in SCR 4 mediates cell adhesion through binding to complement receptor 3 (CR3), an αM integrin. A binding site for C‐reactive protein (CRP), has been localized to the region spanning SCRs 7–11 (blue; in brackets), and a binding site for the Streptococcus pneumoniae surface protein β lies within a domain encompassing SCRs 8–11 (not indicated). Low‐ and high‐affinity adrenomedullin (AM) binding sites have been mapped to SCRs 8–11 and SCRs 15–20 respectively. Three heparin binding sites have been localized to SCRs 7, 12–14, and 20. Also located within SCR 20 is a sialic acid binding site (not indicated). The five related members of the CFH gene family are represented by five separate genes (gene symbols CFHR1–5) all of which reside within the regulators of complement activation (RCA) gene cluster on chromosome 1q32. Each of the 5 CFH‐related genes contains a subset of the 20 CFH SCR domains (see Zipfel, et al., 2002 Citation33 for review). The amino acid sequence homologies of CFHR1–5 SCRs to their CFH counterparts are shown as a percentage in white numerals above each SCR. With the exception of CFHR4 where SCR 7 is absent, all members of the CFH family contain two conserved regions corresponding to SCRs 6–7 and 19–20 of CFH (highlighted in red), and no members contain domains corresponding to SCRs 1–4 of CFH. Based upon their close structural relationships, it may be predicted that the five CFH‐related family members also share functional similarities with CFH.
Although our understanding of nucleotide polymorphisms in loci associated with AMD has increased substantially, especially in the case of CFH, characterization of the disease‐associated CFH risk and protective haplotypes has not yet been assessed fully. In this study, we identify AMD‐associated haplotypes spanning CFH and CFHR1–5. We define a large deletion encompassing two CFH‐related genes—CFHR1 and CFHR3—that further refines our understanding of the protection conferred by the CFH haplotypes identified previously Citation9. We also determine the frequency of a major CFH risk allele (T1277C which encodes Y402H) within numerous human populations. Based on these analyses, we propose a hypothesis to explain the evolution of genetic variation in the CFH locus, with a focus on its association with the development of AMD.
Supplemental data (figures and tables) can be found in the online version of this paper.
Key messages
A large, common deletion that encompasses both the complement factor H‐related 1 (CFHR1) and complement factor H‐related 3 (CFHR3) genes is observed in 1.1% of AMD cases and 5.7% of matched controls (chi‐square = 32.8; P = 1.6 E‐09), indicating that the absence of one or both of these two genes may account for the protective effects conferred by some CFH haplotypes.
The CFHR1/CFHR3 deletion is commonly found in many different populations in the world—an indication of its ancient origin.
The extremely low, practically nonexistent, levels of CFHR1 and CFHR3 transcription by the ocular retinal pigmented epithelium/choroid complex and neural retina suggest that functions ascribed to the respective proteins are mediated through hepatic synthesis and subsequent release into the circulation.
Materials and methods
Patients and donors
Three independent cohorts of AMD cases and AMD‐unaffected, age‐matched controls were used for this study. Individuals in two cohorts were of European‐American descent, and those of the third cohort of Finnish decent; all individuals within each cohort were over the age of 60, unrelated and matched for age and ethnicity. All patients were enrolled under Institutional Review Board‐approved protocols. These groups consisted of 583 individuals with clinically documented AMD and 370 control individuals from the University of Iowa; 635 patients with clinically documented AMD and 357 controls from Columbia University; and 46 patients with clinically documented AMD and 33 unrelated controls from Helsinki University. Patient's eyes were examined and photographed by trained ophthalmologists. Stereo fundus photographs were graded according to standardized classification systems Citation36, Citation37 as described previously Citation9, Citation27. Fundi and/or posterior poles of human donor eyes were graded using the same criteria.
Population frequencies of the CFH Y402H variant and the CFHR1/CFHR3 deletion were assessed using samples from the Human Genome Diversity Panel, which is composed of 1064 individuals from 47 defined ethnic groups Citation38.
Polymerase chain reaction (PCR)
Total genomic DNA was prepared from blood using a QIAamp DNA Blood Kit (Qiagen, Valencia, CA). Identification of CFHR1–5 DNAs was made using polymerase chain reaction (PCR). PCR reactions utilized 10 ng human genomic DNA in the manufacturer's 1X NH4 buffer, 2.5 mM MgCl2, 1 mM deoxynucleotide triphosphate (dNTP) mix, and 10–25 µM of the respective primer pairs. All PCR reactions were performed in a Hybaid thermocycler. Reaction parameters were one cycle at 94°C for 3 minutes; 40 cycles at 94°C for 45 sec; 55.3°C (CFH)/55.3°C (CFHR1)/49.4°C (CFHR2)/50°C (CFHR3)/49.4°C (CFHR4)/49.4°C (CFHR5) for 1 min; 72°C for 1 min; and one cycle at 72°C for 3 min. The PCR products were run on 2% agarose gels and imaged using a Gel Doc 2000™ Documentation System.
Mutation screening and analysis
CFH, CFHR1 and CFHR3 were assessed using single‐strand confirmation polymorphism (SSCP) analyses, denaturing high‐performance liquid chromatography (DHPLC) and direct sequencing as described previously Citation9. Primers for SSCP, DNA PCR amplification and DNA sequencing analyses (Supplemental Table I) were designed to amplify each exon and its adjacent intronic regions using MacVector software (San Diego, CA). Statistical analyses, including chi‐square and Fisher's exact tests, were performed as described previously Citation13.
Haplotype network analysis
Haplotypes were inferred using PHASE 2.1 Citation39, Citation40 choosing the best fit to the European American data as source input for the network. Rare haplotypes (representing less than 0.5% of those inferred) were discarded before input into Network 4.2 (Fluxus Engineering, see: http://www.fluxus‐engineering.com/sharenet.htm). Ancestral sequences were chosen by comparison of the Human May 2004 assembly (hg 17, Build 35) at University of California Santa Cruz (http://genome.ucsc.edu) with November 2003 Chimpanzee sequence. A reduced median network Citation41 was visualized first, followed by a median joining network Citation42 and a median joining network with star contraction Citation43. Each network produced similar findings for the ancestral haplotype and distance from protective and susceptible haplotypes.
Western blot analyses
Human serum samples were collected in red‐topped tubes, centrifuged, processed within 1 hour of collection and stored at −80°C. Serum samples from 65 Iowan and 79 Finnish patients were diluted to a final dilution of 1:200. A total of 4 µL of diluted serum was loaded on 4%–20% gradient gels and the proteins separated by one‐dimensional sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) under nonreducing conditions. Proteins were transferred to a nitrocellulose membrane (Bio‐Rad, Hercules, CA). The membranes were blocked in phosphate buffered saline (PBS) buffer containing 5% nonfat milk for 60 min at room temperature. Polyclonal goat anti‐factor H antiserum (Calbiochem‐Novabiochem Corp. San Diego, California, USA) was diluted in blocking buffer at a concentration of 1:2000. After incubation at 4°C for 16 hours, the membranes were washed four times in PBS followed by incubation with rabbit polyclonal anti‐goat immunoglobulin G conjugated to horse peroxidase (IgG HRP; Abcam, Cambridge, MA) in blocking buffer at a dilution of 1:50000 for 40 min at room temperature. The membrane was washed four times with Tris‐buffered saline containing 0.05% Tween 20 (TBST) prior to developing with Supersignal Femto Chemiluminescent Substrate (Pierce, Rockford, IL).
Human donor eyes
Human donor eyes and livers were obtained from the Iowa Lions Eye Bank within 5 hours of death. Total RNA for RT‐PCR and quantitative polymerase chain reaction (QPCR) analyses was isolated from the retinal pigmented epithelium (RPE)/choroid and retinal punches of eight donors using Qiagen RNeasy mini‐preps (Qiagen, Valencia, CA) according to the manufacturer's suggested protocol. Contaminating DNA was digested with RNAse‐free DNAse and the RNA was repurified using a second RNeasy mini‐column. RNA integrity was assessed by microchannel electrophoresis on an Agilent Bioanalyzer (Agilent Technologies, Inc., Palo Alto, CA).
Quantitative RT‐PCR
Real‐time quantitative polymerase chain reaction (QPCR) was carried out using the SYBR Green method Citation44. Gene‐specific primers were designed using Beacon Design 4.0 (Premier Biosoft International, Palo Alto, CA); the sequences of the PCR primers used are listed in Supplemental Table I.
cDNA was synthesized from 150–640 ng of DNAse‐treated total RNA using the iScript cDNA Synthesis Kit (Bio‐Rad Laboratories). Aliquots representing 1/25th of the resulting cDNA were employed in triplicate 20 µL PCR reactions using the following reaction conditions: 50 mM KCl, 10 mM Tris‐HCl (pH 9.0 at 25°C), 2.5 mM MgCl2, 0.1% Triton X‐100, 0.2 mM dNTPs, 0.20 units Platinum Taq DNA polymerase, 1X SYBR Green (Invitrogen Corp., Carlsbad, CA), and 500 nM sense and anti‐sense gene‐specific primers. Amplification was carried out using the following temperature profile: 3 min at 94°C followed by 45 cycles of 15 sec at 93°C, 15 sec at 56°C, 90 sec at 72°C, 20 sec at 78°C and 20 sec at 82°C. Real‐time fluorescence readings were made at 72°C, 78°C, and 82°C. The specificity of amplification was ascertained by melting profile analysis and gel electrophoresis. For each of the CFH gene family members the analysis was carried out using the measurements made at 78°C. The housekeeping genes (GAPDH, GPI, HMBS, and HPRT1) were quantified using the 82°C measurements. The efficiency of the PCR reaction was determined by standard curve analysis using serial dilutions of cDNA prepared from total human liver RNA (Origene Technologies, Inc., Rockville, MD), and the level of expression was described by the equation: amount = 1/(1+E)Ct, where E is the efficiency and Ct is the PCR cycle number at which the fluorescent signal crosses a predetermined threshold while still undergoing exponential amplification. For each primer set, the threshold was set proportionally to the size of the amplicon to adjust for differences in signal intensities due to product length. The CFH gene family data was normalized to the geometric mean of two ‘housekeeping’ genes (glyceraldehyde phosphate dehydrogenase (GAPDH) and glucose phosphate isomerase (GPI)) whose expression levels were empirically determined to remain constant amongst all tissue types and donors used in this study Citation45.
Results
Absence of CFHR1 protein in sera
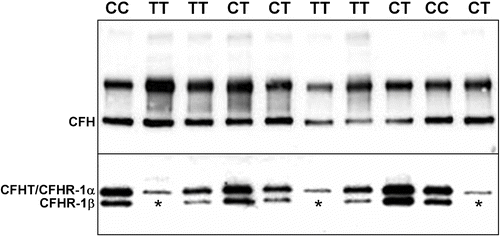
CFHR1 is an abundant plasma protein Citation33, Citation46 that circulates at relatively high concentrations (40 µg/mL). Initial immunoblot analyses of anti‐human CFH antibody‐reactive protein bands in sera obtained from 25 individuals in the Iowa AMD study cohort revealed that CFHR1 (as monitored by reactivity with its glycosylated isoform, CFHR1β) was not detectable in three samples, each of which was derived from control subjects without AMD (two were homozygous for the CFH 1277T allele, and the third was heterozygous at this position) (Figure ). Based on these data, CFHR1‐specific primers were designed and utilized (Supplemental Table I) to assess whether the CFHR1 gene is present in DNA obtained from the same individuals. This analysis indicated that the CFHR1 gene was deleted in the subjects lacking immunoreactive serum CFHR1 protein.
Figure 2 A representative Western blot of serum proteins from 10 of 144 individuals analyzed by nonreducing SDS‐PAGE and chemiluminescent immunoblotting. Upper panel: CFH is visualized as the faster migrating of the two immunoreactive bands (∼150 kDa; identified as CFH by co‐migration with purified CFH; not shown) in the high molecular weight region of the gel/blot. The nature of the higher molecular weight CFH antibody‐reactive band was not determined. The blot depicted was exposed to film for 3 seconds. Lower panel: CFHR1β, a glycosylated isoform of CFHR1, can be distinguished from CFHR1α and CFHT/FHL‐1 which comigrate following separation of proteins in the lower molecular weight region of the gel/blot. No immunoreactive CFHR1β is present in the serum of three patients (lanes 2, 6 and 10), two unaffected patients possessing 1277TT genotypes (protective CFH H4 haplotype) and one patient with a 1277CT genotype, respectively. Subsequent SSCP analysis and direct DNA sequencing showed that all three individuals have both a homozygous deletion of the CFHR1 and CFHR3 genes. The blot was exposed to film for 7 minutes.
Identification of a common deletion spanning the CFHR1 and CFHR3 genes
SSCP analysis and direct DNA sequencing was used to determine the frequency of the homozygous deletion of the CFHR1 gene in 953 additional individuals in the Iowa cohort (Supplemental Table II). Homozygous deletions were documented in 2.7% of this cohort. Using a gene‐specific PCR assay, the homozygous CFHR1 deletion was found in 3.0% of study subjects in the cohort from Columbia University comprised of 992 individuals (Supplemental Table II).
To determine whether the deletion was associated with AMD, we examined the distribution of the deletion homozygotes in the 576 AMD cases and 352 controls from the Iowa cohort (Table ). Deletion homozygotes represented 4.9% of the controls and 1.2% of the cases. This analysis was replicated on a cohort of individuals from Columbia University; 6.7% of the controls and 0.94% of the cases were deletion homozygotes in this cohort. The combined data set demonstrated a highly significant protective effect of the CFHR1 deletion for AMD (chi‐square = 32.8, P = 1.6 E‐09) with an odds ratio of 0.19 (95% CI: 0.099–0.37).
Table I. FHR1 deletion frequencies (cases and controls).
Although individuals heterozygous for the CFHR1 deletion could not be assessed directly in these analyses, we could reliably score most of the samples through examination of their DNA sequence traces (data not shown). These data showed that CFHR1 deletion heterozygotes were less common in cases (17%) than in controls (22%), indicating that the deletion in the heterozygous state may also be somewhat protective.
Primers specific for the CFHR2, CFHR3, CFHR4 and CFHR5 genes (Supplemental Table I) were designed to further characterize the extent of the deletion in the RCA locus. The PCR analyses revealed that the deletion includes the CFHR3 gene (based on assessment of exon 3), but does not include the CFHR2, CFHR4 or CFHR5 genes (Supplemental Figure 1).
Examination of Western blots of serum from an additional 40 Iowa patients revealed that all 10 of the delCFHR1/CFHR3 homozygotes had no detectable CFHR1 protein, whereas the remaining 30 serum samples contained variable amounts of protein (data not shown). These analyses confirmed that delCFHR1/CFHR3 homozygotes lack the CFHR1 gene as well as the corresponding protein. Analysis of a Finnish cohort using Western analysis showed that two controls (2.5%; both CFH 1277TT homozygotes) lacked CFHR1‐reactive bands (Supplemental Table III). CFHR1‐immunoreactive bands were noted in 83.5% of the samples, whereas 13.8% of the samples (all TT and TC at CFH position 1277) exhibited fainter bands presumably derived from CFHR1 heterozygotes. All of the CC homozygotes at CFH position 1277 exhibited robust immunoreactivity. The frequencies of robust, faint and nonexistent immunoreactive CFHR1 bands closely matched expected Hardy‐Weinberg equilibrium predictions for this sample set.
Haplotype analysis of the CFHR1/3 deletion
Analysis of the combined data set was performed to determine what CFH haplotype(s) contained the delCFHR1/CFHR3 allele. The deletion was rarely found on the haplotype tagged by the major CFH risk allele, 402H (Supplemental Table IV). Approximately 63% of CFHR1/CFHR3 deletions lie on the previously identified H4 protective CFH haplotype Citation9; however, not every delCFHR1/CFHR3 chromosome was associated with H4. The protection conferred by delCFHR1/CFHR3 homozygosity was stronger than that of H4 homozygotes (haplotypes for all of the Iowa delCFHR1/CFHR3 homozygotes across the CFH, CFHR1 and CFHR3 genes are depicted in Supplemental Table V). We conclude from these data that some of the previously described protection associated with the CFH H4 haplotype is due to the delCFHR1/CFHR3.
Ocular expression of CFH, CFHR1 and CFHR3
To determine whether CFHR1 and/or CFHR3 transcripts are expressed in the RPE/choroid or neural retina, cDNA was synthesized from total RNA obtained from eight donors. Based upon SSCP analysis, two of these were confirmed as delCFHR1/CFHR3 homozygotes, and the remaining six carried at least one CFHR1 and one CFHR3 allele. Triplicate aliquots were subjected to quantitative real‐time PCR using primers specific for full‐length CFH, truncated CFHT, CFHR1 and CFHR3 (see Supplemental Table I).
No expression of CFHR1 or CFHR3 above background was detected in seven of the eight RPE/choroid samples (Table ). In four of the donors with at least one CFHR1 allele, extremely low expression was detected in the retina, but at levels >250,000‐fold lower than in liver controls. No expression of CFHR1 in the retina was found in the two delCFHR1/CFHR3 homozygotes.
Table II. Ocular CFH, CFHT, CFHR1 and CFHR3 gene expression.
In contrast, mean CFH expression by the RPE/choroid in this group was only 10‐fold below that of liver. CFH expression in the retina was nearly 300‐fold lower than in the RPE/choroid, a level that could be attributable to residual RPE cells that remain adherent to the retina after dissection (Table ). On average, the ratio of full‐length to truncated CFH mRNA was similar in both RPE/choroid and liver (1.5+/−0.4) for those donors with at least one copy of CFHR1/CFHR3. In the two delCFHR1/CFHR3 homozygotes the CFHv1/CFHv2 ratios were somewhat higher (2.2 and 3.7).
Haplotype network analysis
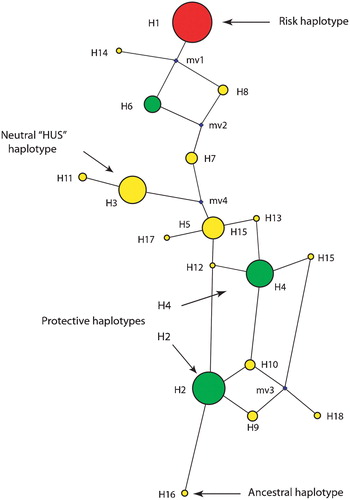
To analyze the relationship of the CFH region haplotypes, including the haplotype(s) associated with the CFHR1/3 deletion, haplotype network analysis was performed (Figure ). This method determines the most likely pathway for the evolution of the haplotypes and displays the predicted haplotype network. A circle that is proportional in size to its abundance depicts the estimated frequency of each haplotype. Haplotype network analysis of the variants in the CFH region revealed that the protective H4 and H5 haplotypes are most closely related to the sequence found in the chimpanzee, and therefore closer to the ancestral state. Haplotypes determined to be neutral in regards to their risk for AMD, as well as the haplotype that confers risk for atypical hemolytic uremic syndrome (aHUS) are more divergent from the ancestral haplotype. The haplotype containing a major CFH risk allele—1277C which encodes 402H—is the most divergent.
Figure 3 A haplotype network resulting from the analysis of variants in theCFH locus is shown. The size of the sphere is proportional to the estimated frequency of the haplotype. Haplotypes adjacent to each other are predicted to arise from a single mutational step. The small spheres represent nodes that were not observed, but are predicted. The 402H haplotype is depicted, along with the two major protective haplotypes, and the haplotypes that are neutral for AMD risk.
Population frequencies of CFH Y402H and deletion alleles
Given the divergence of the CFH T1277C‐containing haplotype, we determined the distribution of this allele in the Human Genome Diversity Panel (HGDP), a set of 1064 DNA samples from 47 defined populations. The CFH 1277C allele occurred at a frequency of 35% in both African and European populations, 25%–30% in North African and Middle Eastern populations, 10%–15% in Asian populations and 0%–5% in Native American populations (Table ). We found extremely high frequencies in the population from Orkney Island (in Scotland) and in Biaka pygmies from the Central African Republic (53%–69%) (data not shown).
Table III. Population frequencies of CFH T1277C and delCFHR1/CFHR3.
The delCFHR1 allele also displays considerable variation between racial/ethnic groups. Deletion homozygotes are most common in African Americans (16%), less common in Hispanics (6.8%) and least common in European Americans (4.7%) (Table ). Analysis of the African samples in the HGDP shows the deletion to be frequent in many different African populations (Table ). The high frequency of both the CFH T1277 allele and the delCFHR1 allele in African populations suggests these alleles have an ancient origin. The high frequency of the deletion in African Americans may explain the documented low frequency of late‐onset AMD in this ethnic group, compared to the Caucasian population Citation47.
Discussion
Recently, our group and several others reported that polymorphisms in CFH, the gene encoding the principal soluble inhibitor of the alternative pathway of the complement cascade, are a major susceptibility factor for the development of AMD Citation9–12. A major ‘risk’ CFH haplotype that confers increased susceptibility to AMD, and two ‘protective’ haplotypes that confer a decreased risk, were identified Citation9.
In the process of serological screening, we noted that another member of the factor H family, CFH‐related protein 1 (CFHR1), as identified by its glycosylated isoform CFHR1β, was not detected in the serum of approximately 3% of individuals. We also noted that most individuals who lacked serum CFHR1 were homozygous for the normal nucleotide T at position 1277 of the CFH gene (which encodes the CFH402Y isoform) and showed no evidence of AMD. Based upon these data, and the knowledge that the CFH‐related genes share numerous regions of homology with CFH, we examined the genetic variation in the RCA locus that includes CFH and the five CFH‐related genes.
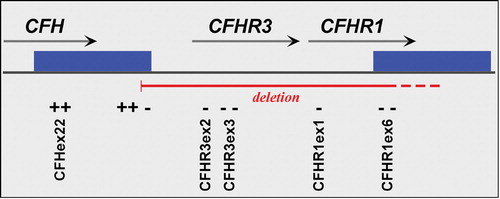
This analysis revealed a relatively common deletion encompassing the entire CFHR1 and CFHR3 genes. Although the precise breakpoints of the deletion were not determined, it includes DNA between the 3' end of exon 22 of CFH and the 5' end of exon 1 of the complement factor H‐related 4 (CFHR4) gene. The breakpoints were further refined using PCR‐specific primers and by chromosome walking (Figure , and data not shown). Hughes and colleagues recently identified the same deletion, although their conclusion that the deletion is exclusively found on a single haplotype (referred to as H5) differs from the data presented here Citation48. Another recent study concluded that the functionally significant CFH S1191L/V1197A mutations in a patient with atypical hemolytic syndrome were caused by de novo gene conversion between CFH exon 22 and homologous sequences in exon 6 of CFHR1Citation49. We suggest that such cases of gene conversion may be more likely to occur when CFH and CFHR1 are in closer proximity, such as they are on CFHR1/CFHR3‐deleted chromosomes.
Figure 4 A schematic map of that portion of the RCA cluster on 1q31 showing the relative positions of theCFH, CFHR1 and CFHR3 genes. The solid green boxes represent a segmental duplication of 28 kb that occurred within this locus. The primers employed to define the CFHR1/CFHR3 deletion boundaries are shown in Supplemental Table I; ‘+’ indicates products that amplify in deletion homozygotes, and ‘–’ regions that fail to amplify in deletion homozygotes.
Immunoblot analyses of sera from individuals homozygous for the CFHR1/CFHR3 deletion confirmed the complete absence of the CFHR1 protein. This observation is consistent with a previous report showing the absence of CFHR1 in the sera of 4.4% of a healthy Caucasian population that was attributed to homozygosity for a null allele in CFHR1Citation50. Two more recent studies, which showed that large deletion variants in the human genome can account for a significant proportion of phenotypic variation, also identified a deletion in CFHR1Citation51, Citation52. 63% of the delCFHR1/CFHR3 alleles are associated with the previously identified CFH H4 haplotype that confers protection against AMD Citation9. This protection may be conferred by the absence of one or both of the CFHR proteins, or by altered function of the specific CFH haplotype(s) with which the deletion is in linkage disequilibrium. The homozygous delCFHR1/CFHR3 is rarely present in individuals affected with AMD (observed in 1.1% of total cases; see Supplemental Table VI). Homozygous deletions were apparent in only 3 individuals with late‐stage choroidal neovascularization (CNV), and in 11 subjects with clinical diagnoses of early AMD. Thus, the homozygous deletion appears to be associated with almost complete protection from late‐stage AMD.
Both CFHR1 and CFHR3 contain SCRs that exhibit significant sequence homology with CFH SCRs 18–20 and 6–7, respectively (Figure ). SCRs 6–7 contain binding sites for heparin, C‐reactive protein, and several bacterial species. This domain also contains the Y402H variant of CFH that confers risk for AMD. The C‐terminal domain of CFH, comprised of SCRs 18–20, has been characterized as mediating target recognition and self/non‐self discrimination in the fluid phase. Function‐blocking antibodies directed against the C‐terminus (SCR20) block interaction of CFH with C3b, C3d, heparin, and binding to endothelial cells Citation53. Mutations in SCR 20 have been shown to result in small vessel thrombosis (microangiopathy), thrombocytopenia, and acute renal failure that are characteristic of aHUS Citation54–56. Although the functional properties of CFHR1 and CFHR3 proteins are not fully defined, these proteins lack the complement regulatory activities ascribed to CFH SCRs 1–4, but retain some of the functions associated with other homologous CFH domains. For example, they bind C3b and C3d, but not C3. Moreover, CFHR3 binds heparin Citation57, and its interaction with opsonized pneumococci can be inhibited by heparin Citation58. Since CFHR1 and CFHR3 both retain a C‐terminal C3 binding domain, but lack the N‐terminal domain responsible for inactivation of C3b, it is reasonable to suggest that they may function as competitive inhibitors of CFH, thus interfering with CFH binding and thereby having a negative effect on complement regulation. In such a model, the deletion of the CFHR1 and CFHR3 genes would lead to enhanced complement inhibitory activity by CFH and, therefore, reduced overall activation of the alternative pathway. Simply put, CFH may be more active in the absence of CFHR1 and CFHR3. In the context of AMD, this may be interpreted as protective if AMD susceptibility, as now believed, is influenced by dysregulation of complement activation via the alternative pathway.
Although the liver is responsible for the synthesis of most circulating CFH, our recent studies show that the eye is also a potential source of CFH, as well as a number of other complement components and complement regulators Citation9. CFH, but not CFHR1 or CFHR3, transcripts can be detected by QPCR in the RPE/choroid complex and neural retina, the tissues most affected in AMD. The absence of CFHR1 and CFHR3 transcription suggests that functions ascribed to the respective proteins may be absent in these tissues or are mediated by systemic CFHRs produced in the liver.
The RCA locus displays a complicated evolutionary and genetic history. The CFH‐related genes were formed by gene duplication and several very large segmental duplications are present in this region. Both T1277C and the CFHR1/CFHR3 deletion are frequent in multiple African populations as well as in European and Asian populations. This result is consistent with the proposition that these variants were selected for and have been maintained in the population by balancing selection. Similarly, the CFH haplotype network analysis demonstrates a diverse set of CFH haplotypes, suggesting that there may be an advantage to maintaining population diversity in the regulation and specificity of the complement response.
The high frequencies of these two alleles in different African populations also suggest that they have an ancient origin. Although we have considered the possibility that the deletion is recurrent, this is unlikely due to the high degree of linkage disequilibrium (LD) with flanking markers. The LD would decay rapidly if a high degree of recurrence existed. We cannot, however, rule out the possibility of a low rate of recurrence of the deletion, nor that duplication alleles with an extra copy of CFHR1/3 may exist.
The population genetic implications of the multiple protective and risk alleles in CFH region are consistent with an important role for the alternative pathway of the complement cascade in pathogen response. In other words, alleles with altered functionality may be maintained to protect against different pathogens. Robust complement activity directed against specific pathogens is desirable; however, it may lead to a higher risk of inflammation‐related, immune response disorders, particularly those with a late onset, which are not subject to selection. Similar diversity is found in other immune function genes, including MHC genes, natural killer receptor genes, immunoglobulin genes and T cell receptor genes.
Identification of a probable role for no CFH in the pathogenetics of AMD has been a major step forward in unraveling the causes of this complex disorder. This finding, together with the subsequent discovery of AMD‐associated variants in two related complement genes (BF and C2) Citation13, Citation26 provide additional compelling evidence that the innate immune system and, more specifically, dysregulation of the alternative pathway of complement, play a central role in the pathobiology of AMD. However, it is not yet apparent how protein isoforms encoded by major AMD risk‐conferring gene variants mediate the molecular and cellular changes leading to AMD. One attractive scenario is that the inflammation triggered by exposure to infectious agents, or some other as yet undiscovered triggering event, in genetically susceptible individuals leads to the sustained activation of complement, drusen formation and, eventually, the development of AMD. This scenario is consistent with the consequences of complement regulatory dysfunction in several rare kidney diseases, including membranoproliferative glomerulonephritis and atypical HUS Citation59, Citation60.
In conclusion, we have demonstrated that there is a common and widespread deletion within the RCA locus that encompasses the CFHR1 and CFHR3 genes. The deletion is commonly found in many different populations in the world—an indication of its ancient origin. The deletion allele is in high linkage disequilibrium with variants in CFH, and the CFH 1277T allele is found on the same haplotype as the deletion with 95% frequency. Those individuals who are homozygous for the CFHR1/CFHR3 deletion and, therefore, do not express the respective proteins, are highly protected from developing AMD. Conversely, it appears that expression of the CFHR1 and CFHR3 proteins, in conjunction with other risk‐conferring CFH isoforms, plays a permissive role in the development of AMD and, perhaps, other age‐related diseases.
Acknowledgements
The authors are grateful to Drs. Amir Arbisser, Lisa Arbisser, Irene Barbazetto, Dean Bok, Peter Dudley, Peter Gouras, Jamal Hoballah, Brian Martin, Patrick Johnson, Tim Johnson, Stephen Russell, Richard Smith, Stacy Thompson, and Thomas Weingeist, for discussions and assistance in recruiting patients. We sincerely appreciate the continued support and technical assistance of Julie Donahue, Kadia Bundu, Sankar Baruah, Christy Curletti, Tiia Falk, Lisa Hardisty, Lindsey Koele, Sheri McCormick, Norma Miller, Katie Peterson, Jake Requard, Taleb Salameh, Kim Silva, Kevin Stichter, Andrew Taiber, Richard Winstrom, Rachel Wolf and the staffs of the Iowa Lions Eye Bank and the Oregon Lions Eye Bank. We are also extremely grateful to our study participants and to all those individuals and families who unselfishly donated their eyes or those of their loved ones to this research program.
This project has been supported by NIH R01 EY11515 (GH), R24 EY017404 (GH, RA, LJ, DA), R01 EY13435 (RA), R01 EY11527 (LJ) and R01 EY11521 (DA), DHHS contract #NO1‐CO‐12400 (MD), the National Cancer Institute (MD), the Inframural Research Program of NIH (MD), International Retina Research Foundation (GH), Foundation Fighting Blindness (RA), Ruth and Milton Steinbach Fund (GH, RA), New York Community Trust (RTS), Wallach Foundation (RA, GRB), Elyachar Foundation (RA, GRB), Kaplen Foundation (RA, GRB), Widgeon Point Charitable Foundation (RA, JEM), Macula Vision Research Foundation (RA) and unrestricted grants to the University of Iowa and Columbia University from Research to Prevent Blindness, Inc. GSH holds honorary professorships in the School of Medicine, Queen's University, Belfast and the Shandong Eye Institute, Qingdao, China.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Web resources
Supplementary Material
Download PDF (88.3 KB)References
- Klein R., Peto T., Bird A., Vannewkirk M. R. The epidemiology of age‐related macular degeneration. Am J Ophthalmol 2004; 137: 486–95
- van Leeuwen R., Klaver C. C., Vingerling J. R., Hofman A., de Jong P. T. Epidemiology of age‐related maculopathy: a review. Eur J Epidemiol 2003; 18: 845–54
- Hageman G. S., Luthert P. J., Victor Chong N. H., Johnson L. V., Anderson D. H., Mullins R. F. An integrated hypothesis that considers drusen as biomarkers of immune‐mediated processes at the RPE‐Bruch's membrane interface in aging and age‐related macular degeneration. Prog Retin Eye Res 2001; 20: 705–32
- Bok D. Evidence for an inflammatory process in age‐related macular degeneration gains new support. Proc Natl Acad Sci U S A 2005; 102: 7053–4
- Anderson D., Mullins R., Hageman G., Johnson L. V. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 2002; 134: 411–31
- Donoso L. A., Kim D., Frost A., Callahan A., Hageman G. The role of inflammation in the pathogenesis of age‐related macular degeneration. Surv Ophthalmol 2006; 51: 137–52
- Sivaprasad S., Chong N. V. The complement system and age‐related macular degeneration. Eye 2006; 20: 867–72
- Wiggs J. L. Complement factor H and macular degeneration: the genome yields an important clue. Arch Ophthalmol 2006; 124: 577–8
- Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci U S A 2005; 102: 7227–32
- Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., et al. Complement factor H polymorphism in age‐related macular degeneration. Science 2005; 308: 385–9
- Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., et al. Complement factor H variant increases the risk of age‐related macular degeneration. Science 2005; 308: 419–21
- Edwards A. O., Ritter R 3rd., Abel K. J., Manning A., Panhuysen C., Farrer L. A. Complement factor H polymorphism and age‐related macular degeneration. Science 2005; 308: 421–4
- Gold B., Merriam J. E., Zernant J., Hancox L. S., Taiber A. J., Gehrs K., et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age‐related macular degeneration. Nat Genet 2006; 38: 458–62
- Thakkinstian A., Han P., McEvoy M., Smith W., Hoh J., Magnusson K., et al. Systematic review and meta‐analysis of the association between complementary factor H Y402H polymorphisms and age‐related macular degeneration. Hum Mol Genet 2006; 15: 2784–90
- Li M., Atmaca‐Sonmez P., Othman M., Branham K. E., Khanna R., Wade M. S., et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age‐related macular degeneration. Nat Genet 2006; 38: 1049–54
- Seddon J. M., George S., Rosner B., Klein M. L. CFH gene variant, Y402H, and smoking, body mass index, environmental associations with advanced age‐related macular degeneration. Hum Hered 2006; 61: 157–65
- Despriet D. D., Klaver C. C., Witteman J. C., Bergen A. A., Kardys I., de Maat M. P., et al. Complement factor H polymorphism, complement activators, and risk of age‐related macular degeneration. JAMA 2006; 296: 301–9
- Grassi M. A., Fingert J. H., Scheetz T. E., Roos B. R., Ritch R., West S. K., et al. Ethnic variation in AMD‐associated complement factor H polymorphism p.Tyr402His. Hum Mutat 2006; 27: 921–5
- Kaur I., Hussain A., Hussain N., Das T., Pathangay A., Mathai A., et al. Analysis of CFH, TLR4, and APOE polymorphism in India suggests the Tyr402His variant of CFH to be a global marker for age‐related macular degeneration. Invest Ophthalmol Vis Sci 2006; 47: 3729–35
- Postel E. A., Agarwal A., Caldwell J., Gallins P., Toth C., Schmidt S., et al. Complement factor H increases risk for atrophic age‐related macular degeneration. Ophthalmology 2006; 113: 1504–7
- Schaumberg D. A., Christen W. G., Kozlowski P., Miller D. T., Ridker P. M., Zee R. Y. A prospective assessment of the Y402H variant in complement factor H, genetic variants in C‐reactive protein, and risk of age‐related macular degeneration. Invest Ophthalmol Vis Sci 2006; 47: 2336–40
- Magnusson K. P., Duan S., Sigurdsson H., Petursson H., Yang Z., Zhao Y., et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med 2006; 3: e5
- Sepp T., Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Moore A. T., et al. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Invest Ophthalmol Vis Sci 2006; 47: 536–40
- Souied E. H., Leveziel N., Richard F., Dragon‐Durey M. A., Coscas G., Soubrane G., et al. Y402H complement factor H polymorphism associated with exudative age‐related macular degeneration in the French population. Mol Vis 2005; 11: 1135–40
- Zareparsi S., Branham K. E., Li M., Shah S., Klein R. J., Ott J., et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age‐related macular degeneration. Am J Hum Genet 2005; 77: 149–53
- Maller J., George S., Purcell S., Fagerness J., Altshuler D., Daly M. J., et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age‐related macular degeneration. Nat Genet 2006; 38: 1055–9
- Seitsonen S., Lemmela S., Holopainen J., Tommila P., Ranta P., Kotamies A., et al. Analysis of variants in the complement factor H, the elongation of very long chain fatty acids‐like 4 and the hemicentin 1 genes of age‐related macular degeneration in the Finnish population. Mol Vis 2006; 12: 796–801
- Gotoh N., Yamada R., Hiratani H., Renault V., Kuroiwa S., Monet M., et al. No association between complement factor H gene polymorphism and exudative age‐related macular degeneration in Japanese. Hum Genet 2006; 120: 139–43
- Okamoto H., Umeda S., Obazawa M., Minami M., Noda T., Mizota A., et al. Complement factor H polymorphisms in Japanese population with age‐related macular degeneration. Mol Vis 2006; 12: 156–8
- Lau L. I., Chen S. J., Cheng C. Y., Yen M. Y., Lee F. L., Lin M. W., et al. Association of the Y402H polymorphism in complement factor H gene and neovascular age‐related macular degeneration in Chinese patients. Invest Ophthalmol Vis Sci 2006; 47: 3242–6
- Sanchez‐Corral P., Perez‐Caballero D., Huarte O., Simckes A. M., Goicoechea E., Lopez‐Trascasa M., et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet 2002; 71: 1285–95
- Rodriguez de Cordoba S., Esparza‐Gordillo J., Goicoechea de Jorge E., Lopez‐Trascasa M., Sanchez‐Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol 2004; 41: 355–67
- Zipfel P. F., Skerka C., Hellwage J., Jokiranta S. T., Meri S., Brade V., et al. Factor H family proteins: on complement, microbes and human diseases. Biochem Soc Trans 2002; 30 Pt 6: 971–8
- Jokiranta T. S., Jaakola V. P., Lehtinen M. J., Parepalo M., Meri S., Goldman A. Structure of complement factor H carboxyl‐terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J 2006; 25: 1784–94
- Friese M. A., Hellwage J., Jokiranta T. S., Meri S., Peter H. H., Eibel H., et al. FHL‐1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol Immunol 1999; 36: 809–18
- Bird A. C., Bressler N. M., Bressler S. B., Chisholm I. H., Coscas G., Davis M. D., et al. An international classification and grading system for age‐related maculopathy and age‐related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol 1995; 39: 367–74
- Klaver C. C., Assink J. J., van Leeuwen R., Wolfs R. C., Vingerling J. R., Stijnen T., et al. Incidence and progression rates of age‐related maculopathy: the Rotterdam Study. Invest Ophthalmol Vis Sci 2001; 42: 2237–41
- Cann H. M., de Toma C., Cazes L., Legrand M. F., Morel V., Piouffre L., et al. A human genome diversity cell line panel. Science 2002; 296: 261–2
- Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing‐data imputation. Am J Hum Genet 2005; 76: 449–62
- Stephens M., Smith N. J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 2001; 68: 978–89
- Bandelt H. J., Forster P., Sykes B. C., Richards M. B. Mitochondrial portraits of human populations using median networks. Genetics 1995; 141: 743–53
- Bandelt H. J., Forster P., Rohl A. Median‐joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48
- Forster P., Torroni A., Renfrew C., Rohl A. Phylogenetic star contraction applied to Asian and Papuan mtDNA evolution. Mol Biol Evol 2001; 18: 1864–81
- Woo T. H., Patel B. K., Cinco M., Smythe L. D., Norris M. A., Symonds M. L., et al. Identification of Leptospira biflexa by real‐time homogeneous detection of rapid cycle PCR product. J Microbiol Methods 1999; 35: 23–30
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., et al. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: RESEARCH0034
- Zipfel P. F., Skerka C. FHL‐1/reconectin: a human complement and immune regulator with cell‐adhesive function. Immunol Today 1999; 20: 135–40
- Klein R., Klein B. E., Knudtson M. D., Wong T. Y., Cotch M. F., Liu K., et al. Prevalence of age‐related macular degeneration in 4 racial/ethnic groups in the multi‐ethnic study of atherosclerosis. Ophthalmology 2006; 113: 373–80
- Hughes A. E., Orr N., Esfandiary H., Diaz‐Torres M., Goodship T., Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age‐related macular degeneration. Nat Genet 2006; 38: 1173–7
- Heinen S., Sanchez‐Corral P., Jackson M. S., Strain L., Goodship J. A., Kemp E. J., et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat 2006; 27: 292–3
- Feifel E., Prodinger W. M., Molgg M., Schwaeble W., Schonitzer D., Koistinen V., et al. Polymorphism and deficiency of human factor H‐related proteins p39 and p37. Immunogenetics 1992; 36: 104–9
- McCarroll S. A., Hadnott T. N., Perry G. H., Sabeti P. C., Zody M. C., Barrett J. C., et al. Common deletion polymorphisms in the human genome. Nat Genet 2006; 38: 86–92
- Conrad D. F., Andrews T. D., Carter N. P., Hurles M. E., Pritchard J. K. A high‐resolution survey of deletion polymorphism in the human genome. Nat Genet 2006; 38: 75–81
- Oppermann M., Manuelian T., Jozsi M., Brandt E., Jokiranta T. S., Heinen S., et al. The C‐terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clin Exp Immunol 2006; 144: 342–52
- Perez‐Caballero D., Gonzalez‐Rubio C., Gallardo M. E., Vera M., Lopez‐Trascasa M., Rodriguez de Cordoba S., et al. Clustering of missense mutations in the C‐terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 2001; 68: 478–84
- Richards A., Buddles M. R., Donne R. L., Kaplan B. S., Kirk E., Venning M. C., et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 2001; 68: 485–90
- Saunders R. E., Goodship T. H., Zipfel P. F., Perkins S. J. An interactive web database of factor H‐associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease‐associated mutations. Hum Mutat 2006; 27: 21–30
- Blackmore T. K., Fischetti V. A., Sadlon T. A., Ward H. M., Gordon D. L. M protein of the group A Streptococcus binds to the seventh short consensus repeat of human complement factor H. Infect Immun 1998; 66: 1427–31
- Hellwage J., Jokiranta T. S., Koistinen V., Vaarala O., Meri S., Zipfel P. F. Functional properties of complement factor H‐related proteins FHR‐3 and FHR‐4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett 1999; 462: 345–52
- Appel G. B., Cook H. T., Hageman G., Jennette J. C., Kashgarian M., Kirschfink M., et al. Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease): An Update. J Am Soc Nephrol 2005; 16: 1392–403
- Abrera‐Abeleda M. A., Nishimura C., Smith J. L., Sethi S., McRae J. L., Murphy B. F., et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet 2006; 43: 582–9