Abstract
Some of the most effective anticancer treatments in clinical use induce DNA damage. The majority of treatments cause severe side effects because they do not specifically target cancer cells but also affect other proliferating cells. Detection of genomic lesions activates the DNA damage response, which determines cell fate according to the extent of damage. If the damage is manageable, the DNA damage response arrests cell cycle progression and induces DNA repair to prevent replication of damaged DNA. If the damage is beyond repair, cells undergo apoptosis. Recently we have shown that the DNA damage response also alerts the innate immune system by inducing the expression of ligands for the activating immune receptor NKG2D. The potential of cancer drugs that target components of the DNA damage response and therapeutic hypotheses to improve current cancer therapies are discussed.
Introduction
Cancer is a complex disease in which cells need to acquire a critical number of genetic changes over many years in order to become cancerous Citation1. The genetic changes can vary greatly between different cancer types and even within single cancer types, in particular in late‐stage cancers, which often display genomic instability. Given the genetic complexity of cancer cells, targeting a single molecule might not lead to the desired effect in all cancer types. However, in recent years a better understanding of the genetic alterations in cancer cells suggests that certain changes, such as rapid proliferation rate and mutations in the DNA repair machinery, might be shared by most cancer types and could serve as potential molecular drug targets.
Many of the effective anticancer treatments in clinical therapies target the DNA by inducing DNA damage Citation2, Citation3. These treatments include ionizing radiation, administration of alkylating agents, DNA topoisomerase inhibitors, platinum compounds, and purine antimetabolites. The key problem with these therapies is that they do not specifically target cancer cells, but also affect other proliferating cells in the body, such as bone marrow cells, epithelial cells of the gastrointestinal tract, and hair follicle cells. Patients under these treatments suffer from severe side effects. In addition, some cancer patients do not respond to certain treatments or they become resistant to the effects of the drugs. Cancer cells can achieve resistance via the active efflux of anticancer drugs by multidrug resistance proteins, modulation of their anticancer activity, increased expression of DNA repair genes, or defects in apoptosis Citation4, Citation5.
In recent years we have gained a better understanding of how cells sense various DNA‐damaging agents, as well as the pathways that mediate the responses to DNA damage. This knowledge is used to design a new generation of therapeutic agents that specifically target the response of cells to DNA damage. Pharmacological inhibition of the DNA damage response is explored to potentiate the efficacy of conventional cancer treatments. In contrast, specific activators promise to replace the current agents used in cancer therapy.
Key messages
Current anticancer agents target DNA nonspecifically resulting in severe side effects in patients.
A better understanding of the DNA damage response in recent years promises new cancer‐specific drugs with better efficacy and fewer side effects.
The finding that the DNA damage response alerts the immune system provides novel strategies for cancer intervention.
DNA damage response
The genome integrity is constantly challenged by environmental genotoxic agents (chemicals, ultraviolet, etc.) and endogenous genotoxic stress (replication, oxidative stress, etc.). The ability to repair damaged DNA is vital for living organisms, as a failure can endanger the survival of the individual cell as well as that of the organism. Cells have evolved surveillance mechanisms to detect genomic insults and to initiate an appropriate response consisting of complex pathways, including sensor, transducer, and effector molecules Citation6, Citation7.
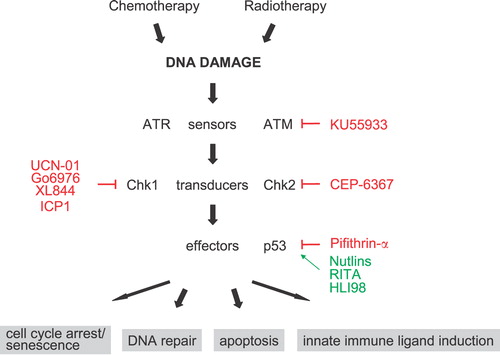
Damages in the DNA lead to the activation of phosphoinositide 3‐kinase‐related protein sensor kinases ATM (ataxia telangiectasia, mutated) and ATR (ATM‐ and Rad3‐related), which initiate the DNA damage response in cooperation with other proteins (Figure ). Double‐strand breaks and changes in chromatin structure are primarily recognized by ATM. In contrast, stalled DNA replication and single‐strand breaks preferentially activate ATR, although both kinases are ultimately activated in response to many genomic lesions. ATM and ATR are central to the DNA damage response and directly activate the Ser/Thr kinases Chk1 and Chk2. These transducer kinases phosphorylate effector proteins such as Brca1, E2F1, Cdc25, and p53 family members, which inhibit cell cycle progression and activate DNA repair systems. If the damage is irreparable, p53 family members induce permanent cell cycle arrest (senescence) or apoptosis. Thus, the DNA damage response preserves genome integrity by either repairing DNA before replication or by inducing apoptosis as a last resort if the damage is too severe to be repaired.
Figure 1 Schematic overview of the DNA damage response. Promising drug targets in the DNA damage response are highlighted. Specific inhibitors are indicated by—|, activators by →.
DNA damage response alerts the immune system
In addition to the known effector functions of the DNA damage response, we recently observed that the expression of ligands for the activating immune receptor NKG2D are upregulated in response to DNA damage Citation8–11. In humans, NKG2D is expressed on natural killer (NK) cells, subsets of CD4+ T cells, natural killer T (NKT) cells, and cytotoxic CD8+ T cells (reviewed in Citation12, Citation13). In contrast, mouse CD8+ T cells express NKG2D upon activation Citation13. Engagement of NKG2D on activated mouse NK cells induces production of inflammatory cytokines, such as interferon‐γ (IFN‐γ). NKG2D is one of the major ‘natural cytotoxicity receptors’ required for NK‐mediated lysis of tumor cell lines in vitro, although other receptors are also involved. On CD8+ T cells, most studies show that NKG2D acts as a costimulatory signal inducing higher levels of T cell immunity Citation13.
NKG2D ligands can be grouped into two families of proteins that are distant relatives of major histocompatibility complex class I molecules: the MIC and RAET1 families Citation12. The MIC gene family members, MICA and MICB, are encoded in the human major histocompatibility complex. No MIC homologs have been found in the mouse genome so far. In contrast, humans and mice share the RAET1 family. The human RAET1 gene family, also called ULBPs, consists of ten genes. The RAET1 proteins are relatively diverse in their amino acid sequence. The mouse Raet1 genes can be further divided into three gene subfamilies: Rae1, H60, and Mult1. These family members are syntenic and structurally related, but share a relatively low degree of homology to each other. The Rae1 gene subfamily consists of several highly related isoforms encoded by different genes. Only one H60 gene and one Mult1 gene have been reported.
The expression of different NKG2D ligands on cells in healthy adults is normally low or not detectable, except on bone marrow cells and mouse thymocytes Citation14, Citation15. In contrast, in diseased cells, such as tumor cells and infected cells, their expression is often upregulated (reviewed in Citation12). MICA and MICB expression is restricted to gastrointestinal epithelial cells in healthy humans, possibly due to the stimulation by the neighboring bacterial flora Citation16. However, expression of MICA and MICB is upregulated on many tumor cell lines and primary tumors of epithelial origin. Similarly, mouse and human RAET1 family members are frequently upregulated on transformed and infected cells, but are undetectable on the surface of normal cells.
A number of reports suggest that ectopic expression of NKG2D ligands in rare tumor cell lines that lack endogenous NKG2D ligands renders the cells sensitive to NK cell‐mediated lysis in vitro and increases their immunogenicity in vivo. In some cases long‐lasting T cell‐mediated immunity against the tumor cells was observed Citation17, Citation18. In addition, administration of a DNA‐vaccine encoding NKG2D ligands and tumor antigens, but not tumor antigens by themselves, induced immune responses that were able to eradicate established tumors Citation19. NKG2D has also been implicated in recent studies to be important in controlling the incidence and progression of cutaneous carcinogenesis and in surveillance of carcinogen‐induced tumors Citation20, Citation21. In humans, tumors may evade NKG2D‐mediated surveillance by shedding soluble MICA and ULBP2, which can be detected in the serum of many cancer patients Citation22–26. The presence of soluble MICA or ULPB2 in the serum correlates with downregulated NKG2D expression and impaired activation of NK and T cells. In summary, a large body of evidence suggests a role for NKG2D in tumor surveillance, but more experimental evidence is needed to provide definite support for this hypothesis.
The restricted expression of NKG2D ligands on transformed or infected cells suggests that changes associated with infection and transformation are sensed by cells and trigger pathways that induce expression of NKG2D ligands on the cell surface. Expression of ligands was suggested to be regulated by loosely defined ‘stress’. Upregulation of MICA or MICB expression on various intestinal epithelial cell lines has been reported to result from the activation of heat shock gene regulatory elements in the promoters of MIC genes. We have not been able to induce the expression of mouse or human Raet1 proteins by various forms of stress, including heat shock, hypoxia, hyperoxia, and serum starvation. These data suggest that different pathways regulate MIC and RAET1 genes Citation8.
Another stress pathway that is relevant to cancer is the DNA damage response Citation27. Recent reports show constitutive activation of the DNA damage response in precancerous and early cancerous lesions of human breast, bladder, lung, and colon, but not in normal tissue Citation28, Citation29. The DNA damage response has also been found to be activated in more than half of the resected human lung and breast tumors. In addition, infection by certain viruses elicits a DNA damage response in cells Citation30. Activation of the DNA damage response could therefore serve as a novel system to sense the disease status of a cell and alert the immune system.
Several new findings now support a connection between the DNA damage response and the immune system. We found that agents damaging the DNA or inhibiting DNA replication induce expression of Raet1 family proteins Citation8. Blocking the function of ATM, ATR, or Chk1 kinases, depending on the nature of the DNA damage, inhibits upregulation of ligands in response to genotoxic agents. Based on the evidence that the DNA damage response is activated in precancerous and cancerous lesions as well as in established tumor cell lines, we tested whether constitutive ligand expression on tumor cells lines depends on the underlying genotoxic stress. We observed that inhibiting ATM, ATR, or Chk1 in tumor cell lines led to a substantial decrease in Rae1 levels at the cell surface Citation8, Citation11. These findings support the idea that persistent genotoxic stress in tumor cell lines maintains constitutive ligand expression and suggest that, depending on the type of DNA damage present in tumor cell lines, either ATM or ATR may be predominantly responsible for maintaining NKG2D ligand expression. In summary, our study suggests that genotoxic stress in tumor cells induces NKG2D ligand expression resulting in the activation of the immune system and may represent an immune barrier in tumorigenesis by recruiting killer cells of the immune systems to lyse diseased cells.
A crucial effector molecule of the DNA damage response is p53. Apoptosis of many tumor types by chemotherapeutic agents and radiation depends on functional p53 Citation31. However, the majority of human cancers display either mutated p53 or defects in p53 functions. An important question therefore is if potential NKG2D‐mediated tumor surveillance in response to DNA damage requires p53 or provides p53‐independent protection against tumor cells. In favor of the latter idea, we found that p53−/− cells are able to upregulate NKG2D ligands in response to DNA damage Citation8. It is plausible that loss of genomic stability often observed in p53‐deficient cells and the resulting genotoxic stress could increase activation of the DNA damage response and upregulate expression of NKG2D ligands on cancer cells.
DNA damage response and targeted drug development
The recent molecular understanding of DNA‐damage‐induced pathways provides new targets for intervention. In the last years efforts have been made to either sensitize cancer cells to DNA‐damaging agents or to develop drugs that selectively induce the DNA damage response in cancer cells (Figure ).
The sensor kinase ATM is one of the components of the DNA damage response that is targeted by specific compounds. The rationale behind the clinical use of ATM inhibitors is that ATM signaling is activated in tumor cells, and that inhibition would render tumor cells hypersensitive to agents that cause double‐strand breaks. A small‐molecule inhibitor of ATM, KU55933, has recently been described, which radio‐ and chemosensitizes cells to ionizing radiation (IR) and a number of DNA damage agents Citation32. Previously, nonspecific ATM/ATR inhibitors, such as wortmannin and caffeine, were tested but were found to be too toxic for clinical exploitation Citation33. DNA‐PKcs, another member of the phosphatidylinositol 3‐kinase‐like kinase family, has also been proposed as a drug target. Several small‐molecule inhibitors of DNA‐PKcs have been developed, including vanillin, OK‐1035, NU7441 and IC87361 Citation34, Citation35. These molecules sensitize cells to radiation and DNA‐damaging agents in vitro and in vivo.
Among the most clinically advanced drugs are Chk1 inhibitors, some of which are tested in phase I/II clinical trials for cancer indications. Chk1 is crucial for normal cell cycle progression and maintaining genome stability in the absence of environmental genotoxic stress Citation36–38. Examination of blastocyst cells derived from Chk1−/− mice, which die early in embryogenesis, shows loss of chromosome integrity and increased apoptosis Citation39. These observations lead to the hypothesis that Chk1 monitors and stabilizes replication forks thereby preventing spontaneous DNA damage during replication Citation36, Citation40–42. However, a Chk1−/− chicken B cell lymphoma cell line is viable, and reduction of Chk1 by siRNA approaches does not induce apoptosis, indicating that Chk1 is not absolutely required for the viability of somatic cells Citation3, Citation38, Citation43. A large body of evidence suggests that inhibiting Chk1 sensitizes tumor cells, particularly those that lack p53, to a broad spectrum of DNA‐damaging agents Citation3, Citation41, Citation43–51. The increased sensitivity of cells lacking p53 to Chk1 inhibitors could have important therapeutic implications, as tumor cells with defects in certain checkpoints might be more responsive to such drugs than normal cells. It remains to be established if proliferating somatic cells are less sensitive to genomic instability caused by Chk1 inhibition than tumor or embryonic cells. No myelosuppression was observed in phase I trials with UCN‐01, a Chk1/PKC inhibitor, suggesting that Chk1 is not essential in somatic cells. Alternatively, it is possible that UCN‐01 only partially inhibits Chk1 in vivo due to its property to get sequestered by serum proteins, which compromises the bioavailability and potency of UCN‐01 Citation52. The results from these ongoing trials will provide important information regarding the potential and toxicity of Chk1 inhibitors, despite the limitation of UCN‐01. It will be of interest to see the clinical results of more specific Chk1 inhibitors, such as SB‐218078, Go6976, XL844, or ICP‐1 Citation53.
In contrast to Chk1, loss or inhibition of Chk2 does not seem to have chemosensitization effects Citation3, Citation54–56. Instead, Chk2‐deficient mice survive higher whole‐body IR doses than wild‐type mice, and several cell types derived from Chk2−/− mice show increased resistance to IR‐induced apoptosis Citation56. Chk2 is required for IR‐induced transcription of several p53 targets, such as the proapoptotic regulator BCL2‐associated X protein (BAX), although it is not understood how Chk2 regulates p53 activity Citation55, Citation57. Chk2 is highly expressed in normal tissues, such as bone marrow cells and epithelial cells of the gastrointestinal tract, which are affected by DNA‐damaging cancer agents Citation58. It is possible therefore that Chk2 is causing some of the toxic side effects of conventional anticancer treatments, and inhibition of Chk2 by specific molecules might reduce damage to normal cells. Hence, inhibition of Chk2 by targeted drugs would not render tumor cells more chemosensitive, but would rather allow widening the therapeutic window of DNA‐damaging agents by reducing their toxic effect on normal tissue. Although Chk2−/− mice do not spontaneously develop tumors, it remains to be seen if inhibiting Chk2 in anticancer treatments will lead to tumor formation by preventing apoptosis of damaged cells. Encouragingly, studies using the p53 inhibitor pifithrin‐α suggest that transient inhibition of the Chk2‐p53 pathway might not have a tumor‐promoting effect Citation55, Citation59. A specific inhibitor like CEP‐6367 will allow testing the potential usefulness of Chk2 inhibition Citation60.
Activation of p53 by nongenotoxic compounds has been proposed as a new therapeutic approach for treatment of tumors. p53 is maintained at low levels in cells mainly by mouse double minute 2 (MDM2)‐mediated degradation Citation61. Thus, small molecules including nutlin‐3 Citation62, RITA Citation63 and HLI98 Citation64, that disrupt the MDM2‐p53 interaction, prevent MDM2‐mediated p53 degradation and restore p53 function in tumors Citation65. These compounds effectively trigger p53 activation and apoptosis in tumor cells that retain normal p53‐mediated apoptosis pathways or cell cycle arrest but not in tumors in which p53‐mediated apoptosis is deregulated Citation66–69. They are also inactive in tumors that harbor mutant p53. A concern with modulators of p53 function is that they activate p53 in normal cells and little is known about the consequences of p53 activation in normal proliferating cells in vivo. In animal xenograft models, nutlins controlled the growth of tumors without apparent toxicity to the normal tissue. Nevertheless, cells derived from thymus, spleen, and epithelium of the small intestine of mice that express hypomorphic levels of MDM2 show elevated levels of apoptosis Citation70. In addition, conditional knockout of MDM2 from specific tissues lead to spontaneous activation of p53 Citation71–73. It has been suggested that nutlins only partially activate p53 because of reversible binding to MDM2 and limited access to cells in the animal, but toxicity in normal tissues may still significantly decrease the therapeutic use of p53 modulators, and further studies are needed to assess their safety for humans.
Conclusion and future directions
During recent years considerable progress has been made in the understanding of the DNA damage response. Specific inhibitors of components of the DNA damage response could selectively increase the efficacy of anticancer treatments that target the DNA by either taking advantage of the DNA repair and cell cycle checkpoint defects in cancer cells or by inhibiting the response of normal cells to DNA damage. The molecular mechanisms leading to chemosensitization of cancer cells are still poorly understood but appear to require inhibition of certain aspects of the DNA damage response, in particular inhibition of DNA repair. A better understanding of pathways triggered in response to DNA damage is going be crucial to evaluate the therapeutic potential of targets in the DNA damage response.
Several proteins of the DNA damage response are currently under investigation with respect to their therapeutic potential. Inhibition of Chk1 might render proliferating cells sensitive to spontaneous DNA damage but could also be toxic to normal proliferating cells, such as bone marrow and some intestinal cells Citation39, Citation74. Inhibition of Chk2 or p53 by small molecules could protect normal cells from cell death during chemotherapy; however, they might still die through mitotic catastrophe. Activation of p53 by MDM2 antagonists could trigger fatal pathologies that include ablation of radiosensitive tissues and profound inhibition of cell proliferation in apoptosis‐resistant tissues as suggested by studies using MDM2‐deficient deficient mice Citation61. More experimental evidence is required to firmly support the feasibility and safety of drugs that target the DNA damage response.
Our recent finding that the DNA damage response alerts the immune system reveals novel strategies for cancer intervention. The efficacy of chemo‐ and radiotherapy depends on the function of p53. However, many cancer patients with advanced tumors have deficiencies in p53 function. It is therefore possible that chemo‐ or radiotherapy‐induced expression of NKG2D ligands on tumor cells, which does not depend on p53, could account for some of the efficacy of these treatments Citation8, Citation66, Citation75. In a number of experimental tumor models it was observed that low doses of certain chemotherapeutic agents enhance the antitumor immune response Citation76. The common high‐dose chemotherapy is immunosuppressive, but believed to be more efficient in killing tumor cells. However, low‐dose chemotherapy was found to be sometimes equal or even superior to high‐dose chemotherapy Citation77. Enhancing NK cell and T cell immunity by for example administering cytokines such as interleukin‐15 in low‐dose chemotherapy or radiotherapy could be an effective strategy to treat cancer. A more detailed molecular understanding of the DNA damage response may aid in the design of novel therapeutic approaches and selective compounds with better efficacy and fewer side effects.
Acknowledgements
I thank Dr David H. Raulet for giving me the opportunity to write this review. I also thank Dr Asja Praetor for reading the manuscript and helpful discussion. This work was supported by grants from the NUS Office of Life Sciences to S. Gasser.
References
- Hahn W. C., Weinberg R. A. Modelling the molecular circuitry of cancer. Nat Rev Cancer 2002; 2: 331–41
- Zhou B. B., Anderson H. J., Roberge M. Targeting DNA checkpoint kinases in cancer therapy. Cancer Biol Ther 2003; 2 Suppl 1: S16–22
- Zhou B. B., Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer 2004; 4: 216–25
- Gottesman M. M., Fojo T., Bates S. E. Multidrug resistance in cancer: role of ATP‐dependent transporters. Nat Rev Cancer 2002; 2: 48–58
- Szakacs G., Paterson J. K., Ludwig J. A., Booth‐Genthe C., Gottesman M. M. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5: 219–34
- Kastan M. B., Bartek J. Cell‐cycle checkpoints and cancer. Nature 2004; 432: 316–23
- Sancar A., Lindsey‐Boltz L. A., Unsal‐Kaccmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85
- Gasser S., Orsulic S., Brown E. J., Raulet D. H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005; 436: 1186–90
- Gasser S., Raulet D. The DNA damage response, immunity and cancer. Semin Cancer Biol 2006; 16: 344–7
- Gasser S., Raulet D. H. The DNA damage response arouses the immune system. Cancer Res 2006; 66: 3959–62
- Gasser S., Raulet D. H. Activation and self‐tolerance of natural killer cells. Immunol Rev 2006; 214: 130–42
- Raulet D. H. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 2003; 3: 781–90
- Jamieson A. M., Diefenbach A., McMahon C. W., Xiong N., Carlyle J. R., Raulet D. H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002; 17: 19–29
- Diefenbach A., Jamieson A. M., Liu S. D., Shastri N., Raulet D. H. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol 2000; 1: 119–26
- Nowbakht P., Ionescu M. C., Rohner A., Kalberer C. P., Rossy E., Mori L., et al. Ligands for natural killer cell‐activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood 2005; 105: 3615–22
- Groh V., Bahram S., Bauer S., Herman A., Beauchamp M., Spies T. Cell stress‐regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A 1996; 93: 12445–50
- Diefenbach A., Jensen E. R., Jamieson A. M., Raulet D. H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001; 413: 165–71
- Hayakawa Y., Kelly J. M., Westwood J., Darcy P. K., Diefenbach A., Raulet D. H., et al. Tumor rejection mediated by NKG2D receptor‐ligand interaction is strictly dependent on perforin. J Immunol 2002; 169: 5377–81
- Zhou H., Luo Y., Kaplan C. D., Kruger J. A., Lee S. H., Xiang R., et al. A DNA‐based cancer vaccine enhances lymphocyte crosstalk by engaging the NKG2D receptor. Blood 2005; 107: 3251–7
- Oppenheim D. E., Roberts S. J., Clarke S. L., Filler R., Lewis J. M., Tigelaar R. E., et al. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol 2005; 6: 928–37
- Smyth M. J., Swann J., Cretney E., Zerafa N., Yokoyama W. M., Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med 2005; 202: 583–8
- Groh V., Wu J., Yee C., Spies T. Tumour‐derived soluble MIC ligands impair expression of NKG2D and T‐cell activation. Nature 2002; 419: 734–8
- Holdenrieder S., Stieber P., Peterfi A., Nagel D., Steinle A., Salih H. R. Soluble MICA in malignant diseases. Int J Cancer 2006; 118: 684–7
- Raffaghello L., Prigione I., Airoldi I., Camoriano M., Levreri I., Gambini C., et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 2004; 6: 558–68
- Salih H. R., Antropius H., Gieseke F., Lutz S. Z., Kanz L., Rammensee H. G., et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003; 102: 1389–96
- Waldhauer I., Steinle A. Proteolytic release of soluble UL16‐binding protein 2 from tumor cells. Cancer Res 2006; 66: 2520–6
- Halazonetis T. D. Constitutively active DNA damage checkpoint pathways as the driving force for the high frequency of p53 mutations in human cancer. DNA Repair (Amst) 2004; 3: 1057–62
- Gorgoulis V. G., Vassiliou L. V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434: 907–13
- Bartkova J., Horejsi Z., Koed K., Kramer A., Tort F., Zieger K., et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005; 434: 864–70
- Weitzman M. D., Carson C. T., Schwartz R. A., Lilley C. E. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 2004; 3: 1165–73
- Wallace‐Brodeur R. R., Lowe S. W. Clinical implications of p53 mutations. Cell Mol Life Sci 1999; 55: 64–75
- Hickson I., Zhao Y., Richardson C. J., Green S. J., Martin N. M., Orr A. I., et al. Identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM. Cancer Res 2004; 64: 9152–9
- Lord C. J., Garrett M. D., Ashworth A. Targeting the double‐strand DNA break repair pathway as a therapeutic strategy. Clin Cancer Res 2006; 12: 4463–8
- Ding J., Miao Z. H., Meng L. H., Geng M. Y. Emerging cancer therapeutic opportunities target DNA‐repair systems. Trends Pharmacol Sci 2006; 27: 338–44
- Madhusudan S., Middleton M. R. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev 2005; 31: 603–17
- Petermann E., Caldecott K. W. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle 2006; 5: 2203–09
- Chen Y., Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair (Amst) 2004; 3: 1025–32
- Canman C. E. Replication checkpoint: preventing mitotic catastrophe. Curr Biol 2001; 11: R121–4
- Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14: 1448–59
- Zachos G., Rainey M. D., Gillespie D. A. Chk1‐dependent S‐M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol 2005; 25: 563–74
- Zachos G., Rainey M. D., Gillespie D. A. Chk1‐deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 2003; 22: 713–23
- Bartek J., Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3: 421–9
- Chen Z., Xiao Z., Chen J., Ng S. C., Sowin T., Sham H., et al. Human Chk1 expression is dispensable for somatic cell death and critical for sustaining G2 DNA damage checkpoint. Mol Cancer Ther 2003; 2: 543–8
- Russell K. J., Wiens L. W., Demers G. W., Galloway D. A., Plon S. E., Groudine M. Abrogation of the G2 checkpoint results in differential radiosensitization of G1 checkpoint‐deficient and G1 checkpoint‐competent cells. Cancer Res 1995; 55: 1639–42
- Shao R. G., Cao C. X., Shimizu T., O'Connor P. M., Kohn K. W., Pommier Y. Abrogation of an S‐phase checkpoint and potentiation of camptothecin cytotoxicity by 7‐hydroxystaurosporine (UCN‐01) in human cancer cell lines, possibly influenced by p53 function. Cancer Res 1997; 57: 4029–35
- Bunch R. T., Eastman A. Enhancement of cisplatin‐induced cytotoxicity by 7‐hydroxystaurosporine (UCN‐01), a new G2‐checkpoint inhibitor. Clin Cancer Res 1996; 2: 791–7
- Hsueh C. T., Kelsen D., Schwartz G. K. UCN‐01 suppresses thymidylate synthase gene expression and enhances 5‐fluorouracil‐induced apoptosis in a sequence‐dependent manner. Clin Cancer Res 1998; 4: 2201–06
- Akinaga S., Nomura K., Gomi K., Okabe M. Enhancement of antitumor activity of mitomycin C in vitro and in vivo by UCN‐01, a selective inhibitor of protein kinase C. Cancer Chemother Pharmacol 1993; 32: 183–9
- Wang H., Wang X., Zhou X. Y., Chen D. J., Li G. C., Iliakis G., et al. Ku affects the ataxia and Rad 3‐related/CHK1‐dependent S phase checkpoint response after camptothecin treatment. Cancer Res 2002; 62: 2483–7
- Koniaras K., Cuddihy A. R., Christopoulos H., Hogg A., O'Connell M. J. Inhibition of Chk1‐dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 2001; 20: 7453–63
- Nghiem P., Park P. K., Kim Y., Vaziri C., Schreiber S. L. ATR inhibition selectively sensitizes G1 checkpoint‐deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A 2001; 98: 9092–7
- Sausville E. A., Arbuck S. G., Messmann R., Headlee D., Bauer K. S., Lush R. M., et al. Phase I trial of 72‐hour continuous infusion UCN‐01 in patients with refractory neoplasms. J Clin Oncol 2001; 19: 2319–33
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004; 3: 513–19
- Jack M. T., Woo R. A., Hirao A., Cheung A., Mak T. W., Lee P. W. Chk2 is dispensable for p53‐mediated G1 arrest but is required for a latent p53‐mediated apoptotic response. Proc Natl Acad Sci U S A 2002; 99: 9825–9
- Hirao A., Cheung A., Duncan G., Girard P. M., Elia A. J., Wakeham A., et al. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)‐dependent and an ATM‐independent manner. Mol Cell Biol 2002; 22: 6521–32
- Takai H., Naka K., Okada Y., Watanabe M., Harada N., Saito S., et al. Chk2‐deficient mice exhibit radioresistance and defective p53‐mediated transcription. EMBO J 2002; 21: 5195–205
- Hirao A., Kong Y. Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., et al. DNA damage‐induced activation of p53 by the checkpoint kinase Chk2. Science 2000; 287: 1824–7
- Lukas C., Bartkova J., Latella L., Falck J., Mailand N., Schroeder T., et al. DNA damage‐activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res 2001; 61: 4990–3
- Komarov P. G., Komarova E. A., Kondratov R. V., Christov‐Tselkov K., Coon J. S., Chernov M. V., et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285: 1733–7
- Sorensen C. S., Syljuasen R. G., Falck J., Schroeder T., Ronnstrand L., Khanna K. K., et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation‐induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3: 247–58
- Ringshausen I., O'Shea C. C., Finch A. J., Swigart L. B., Evan G. I. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10: 501–14
- Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., et al. In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science 2004; 303: 844–8
- Issaeva N., Bozko P., Enge M., Protopopova M., Verhoef L. G., Masucci M., et al. Small molecule RITA binds to p53, blocks p53‐HDM‐2 interaction and activates p53 function in tumors. Nat Med 2004; 10: 1321–8
- Yang Y., Ludwig R. L., Jensen J. P., Pierre S. A., Medaglia M. V., Davydov I. V., et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7: 547–59
- Klein C., Vassilev L. T. Targeting the p53‐MDM2 interaction to treat cancer. Br J Cancer 2004; 91: 1415–19
- Coll‐Mulet L., Iglesias‐Serret D., Santidrian A. F., Cosialls A. M., de Frias M., Castano E., et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B‐cell chronic lymphocytic leukemia cells. Blood 2006; 107: 4109–14
- Carvajal D., Tovar C., Yang H., Vu B. T., Heimbrook D. C., Vassilev L. T. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res 2005; 65: 1918–24
- Tovar C., Rosinski J., Filipovic Z., Higgins B., Kolinsky K., Hilton H., et al. Small‐molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A 2006; 103((6))1888–93
- Secchiero P., Barbarotto E., Tiribelli M., Zerbinati C., di Iasio M. G., Gonelli A., et al. Functional integrity of the p53‐mediated apoptotic pathway induced by the nongenotoxic agent nutlin‐3 in B‐cell chronic lymphocytic leukemia (B‐CLL). Blood 2006; 107: 4122–9
- Mendrysa S. M., McElwee M. K., Michalowski J., O'Leary K. A., Young K. M., Perry M. E. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol 2003; 23: 462–72
- Xiong S., Van Pelt C. S., Elizondo‐Fraire A. C., Liu G., Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci U S A 2006; 103: 3226–31
- Francoz S., Froment P., Bogaerts S., De Clercq S., Maetens M., Doumont G., et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci U S A 2006; 103: 3232–7
- Boesten L. S., Zadelaar S. M., De Clercq S., Francoz S., van Nieuwkoop A., Biessen E. A., et al. Mdm2, but not Mdm4, protects terminally differentiated smooth muscle cells from p53‐mediated caspase‐3‐independent cell death. Cell Death Differ 2006; 13: 2089–98
- Syljuasen R. G., Sorensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25: 3553–62
- Toledo F., Wahl G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev 2006; 6: 909–23
- Zagozdzon R., Golab J. Immunomodulation by anticancer chemotherapy: more is not always better (review). Int J Oncology 2001; 18: 417–24
- DiPaola R. S., Durivage H. J., Kamen B. A. High time for low‐dose prospective clinical trials. Cancer 2003; 98: 1559–61