Abstract
Background. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only proven curative therapy for chronic myeloid leukemia (CML), but lack of human leukocyte antigen (HLA)-matched sibling or unrelated donors has restricted its application. Recently, we developed an effective method for haploidentical allo-HSCT achieving comparable outcomes to HLA-identical transplantation.
Aim. To evaluate the outcomes of CML patients who underwent haploidentical allo-HSCT.
Methods. Ninety-three patients were treated with a modified busulfan(BU)/cyclophosphamide (CY)2 regimen, including antithymocyte globulin followed by unmanipulated blood and marrow transplantation.
Results. Our data showed that the cumulative incidence of acute graft-versus-host disease (GVHD) was 64.52%, and grade III–IV was 26.45%, 61.79% had chronic GVHD, and 28.93% had extensive chronic GVHD. Non-relapse mortality varied at 8.72% (100 days), 20.72% (1 year) and 20.72% (2 years). Probability of 1-year and 4-year leukemia-free survival was similar in chronic phase (CP) 1, CP2/CR2, accelerated phase, and blast crisis patients. Probability of 4-year overall survival varied as 76.5% (CP1), 85.7% (CP2/CR2), 73.3% (accelerated phase), and 61.5% (blast crisis). Multivariate analysis indicated that factors affecting transplantation outcomes were HLA-B+DR mismatches versus others for II–III acute GVHD and III–IV acute GVHD, the stage of disease at transplantation for relapse, and the time from diagnosis to transplantation for leukemia-free survival, overall survival, and transplantation-related mortality. In our protocol, survival of HSCT for advanced CML was similar to stable stage.
Conclusions. For patients lacking an HLA-identical related donor, haploidentical relatives are alternative HSCT donors.
Introduction
Chronic myeloid leukemia (CML) is a common malignant clonal disorder of hematopoietic stem cells. Despite promising advances in CML therapy with the tyrosine kinase inhibitor imatinib mesylate Citation1, Citation2, allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only proven curative therapy for CML, particularly for CML patients in the accelerated phase (AP) or blast crisis (BC) Citation3, Citation4. Transplant from a human leukocyte antigen (HLA)-matched related donor is the primary technique for allogeneic transplantation; however, only 25%–30% of eligible patients have a related donor with suitable or closely matched HLA. For patients without an HLA-matched sibling, bone marrow or umbilical cord from an unrelated donor is often used as a source of stem cells for transplantation Citation5, Citation6. Unfortunately, this therapy may be unavailable due to an unsuccessful donor search or insufficient amount of cord blood cells Citation7, and the alternative is hematopoietic stem cells from an HLA-mismatched family donor Citation8–13. This less than ideal approach, particularly prior to the end of the last decade, was associated with poor engraftment, high risk of early death, and graft-versus-host disease (GVHD). The approach of T cell depletion (TCD) or CD34 cell selection in vitro overcame the HLA barrier but was also challenged with delayed immune reconstitution, infectious complications, and poor survival Citation11, Citation12.
Abbreviations
Recently, we developed a new method for haploidentical transplantation without in vitro T cell depletion Citation14, Citation15. The strategy (the GIAC protocol) applies sequential, in vivo modulation of the recipient, the donor T cell function, and the dose of donor hematopoietic stem cells. The protocol entails the following: donor treatment with granulocyte colony-stimulating factor (G-CSF) to induce donor immunological tolerance; intensified immunological suppression to both promote engraftment and to prevent GVHD; anti-human thymocyte immunoglobulin (ATG) was included for the prophylaxis of GVHD and graft reject; and combination of G-CSF-primed bone marrow harvest and G-CSF-mobilized peripheral blood stem cell harvest as the source of stem cell grafts. Via this GIAC protocol, promising results have been achieved in our institute Citation14, Citation15.
Key messages
Recently, promising results have been achieved in our institute using a new method for haploidentical transplantation without in vitro T cell depletion.
In this study, we found that for chronic myeloid leukemia (CML) patients without a human leukocyte antigen (HLA)-identical sibling donor, a haploidentical family member can serve as an alternative donor.
The optimal time for hematopoietic stem cell transplantation (HSCT) in CML patients from HLA-mismatched family donors needs to be further studied.
In this article we report the outcomes of 93 consecutive patients with CML who underwent transplantation via our new transplant protocol and discuss possible factors that influence the outcomes of the transplantation.
Patients and methods
Patient eligibility
Ninety-three consecutive patients with CML underwent HLA haploidentical allo-HSCT between July 2002 and September 2006 at the Peking University Institute of Hematology. We enrolled patients with CML that were suitable for allo-HSCT and had no HLA-identical related or unrelated donors. Patients were not eligible for HSCT if they had any active infections or severe liver/renal/lung or heart diseases. The Institutional Review Board of Peking University approved this study, and all patients and their donors gave written informed consent.
Details of the patient population and of two groups of patients divided according to the degree of HLA-disparity between recipient and donor (group 1: 16 patients transplanted from donors mismatched for 1 HLA antigens; and group 2: 77 patients transplanted from 2–3 HLA antigens mismatched family members) are given in . Twenty patients received HSCT mismatched for class I (A, B, or A + B) antigens only, seven patients were given HSCT mismatched II class (DR) antigens only, and the remaining 66 patients received HSCT mismatched for both class I and class II antigens ().
Table I. Patients’ characteristics.
Table II. Comparison of patients transplanted with HLA class I, class I + II, or class I + II mismatched donors.
Donor and stem cell harvesting
Familial donors were ranked on the basis of the best HLA match, age (younger preferred), relationship (mother preferred), gender (same preferred), and health status (better preferred). For HLA-A and B, low-resolution DNA techniques were used. High-resolution techniques were used to do class II antigens.
Donors were treated with G-CSF (Filgrastim, Kirin, Japan; 5 µg/kg per day) injected subcutaneously (s.c.) for 5–6 consecutive days. On the fourth day, bone marrow cells were harvested. The target mononuclear cell count (MNC) was 3×108–4×108 cells/kg recipient weight. On the fifth and sixth days, peripheral blood stem cells (PBSCs) were collected with a COBE blood cell separator (Spectra LRS, COBE BCT Inc., Lakewood, CO, USA) at a rate of 80 mL/min from a total blood volume of 10 L. The fresh and unmanipulated bone marrow and PBSCs were infused into the recipient on the day of collection. In instances of incompatibility with the major ABO blood group, red cells were removed from bone marrow cells by density gradient sedimentation with Hespan (B. Braun Medical Inc, Irvine, CA, USA), according to the manufacturer's instructions. Surface markers of the cells in graft were determined by multi-color staining using monoclonal antibodies specific for CD34, CD3, CD4, and CD8 cells, essentially as described by Huang et al. Citation16.
Conditioning regimen
All patients were treated with regimen A as described previously Citation15, which consisted of cytosine arabinoside (4 g/m2/d, intravenous (i.v.)) on day 10 and 9 before transplantation, busulfan (12 mg/kg per os (p.o.) in 12 doses) on day 8, 7, and 6, cyclophosphamide (1.8 g/m2/d, i.v.) on day 5 and 4, semustine (250 mg/kg, i.v.) on day 3, and ATG (2.5 mg/kg/d, i.v. of the Sangstat product) on day 5 to 2.
GVHD prophylaxis
All transplant recipients received cyclosporin A (CsA), mycophenolate mofetil (MMF), and short-term methotrexate (MTX). The dosage of CsA was 2.5 mg/kg/d, i.v. from day 9 before transplantation until bowel function returned to normal. At that point, the patient was switched to oral CsA. MMF was administered orally, 0.5 g every 12 h, from day 9 before transplantation to day 30 after transplantation and tapered half on day +30, discontinued on day +60. The dosage of methotrexate was 15 mg/m2, administered i.v. on day 1, and 10 mg/m2 on days 3, 6, and 11 after transplantation. Whole-blood CsA concentration was monitored weekly using fluorescence polarization immunoassay, and the dosage was adjusted to attain a blood concentration of 150–250 ng/mL. In cases where no evidence of GVHD was detected by days +40 to +50, the CsA dosage was reduced gradually and discontinued around day +180. In cases where GVHD was detected, CsA was continued.
Evaluation of engraftment
Engraftment was defined as the maintenance of an absolute neutrophil count above 0.5×109/L for three consecutive days after the neutrophil nadir. Follow-up assessments were performed by cytogenetic analysis of bone marrow aspiration in the first, second, and third month after transplantation. Chimerism was determined by at least two of the three following methods: DNA-based HLA typing, polymerase chain reaction-based (PCR-based) DNA fingerprinting of short tandem repeat, and chromosomal fluorescent in situ hybridization (FISH).
Chimerism analyses and minimal residual disease monitor
Minimal residual disease (MRD) was evaluated on recipient bone marrow (BM) cells on days +30, +60, +90, +120, +150, +180, and +365 after HSCT by fluorescence in situ hybridization (FISH). MRD was also monitored by Bcr/Abl (quantitative or qualitative polymerase chain reaction, PCR).
Imatinib and donor lymphocyte infusion (DLI)
Before transplantation, 30 patients, including 4 in CP, 7 in AP, and 19 in BC, were treated with imatinib mesylate at 400 mg daily; no significant side effects were noted. Among them, 12 achieved a complete hematologic response, 11 a major hematologic response, 2 a partial response and 5 had no response. Donor lymphocyte infusion (DLI) and imatinib mesylate were given to patients who had a cytogenetic relapse or a hematologic relapse after HSCT, following a trial of immunosuppressant withdrawal. Cytogenetic relapse was defined as Philadelphia (Ph) chromosome reappearance, and hematologic relapse was defined as according to the criteria of the World Health Organization (WHO). In patients without GVHD, G-CSF-mobilized peripheral blood stem cell grafts were used for DLI. The rationale for using growth factor-mobilized peripheral blood stem cells is that G-CSF-primed peripheral blood stem cell harvests contain more CD34+, CD14+, and Th2 cells than non-primed peripheral lymphocyte harvests, which is therapeutically effective in our transplant settings, so the incidence of acute GVHD could be decreased without abrogating the graft-versus-leukemia effect Citation17, Citation18. Otherwise imatinib mesylate was given. The dose of imatinib mesylate was initially 400 mg/day, and was adjusted, when necessary, according to response and tolerance.
Supportive care
All patients were hospitalized in rooms with high-efficiency particle-arresting (HEPA) air filters, and all received antibiotic prophylaxis with oral trimethoprim-sulfamethoxazole, fluconazole, and acyclovir. All blood products were irradiated before infusion. Human immunoglobulin (400 mg/kg) was given i.v. on days 1, 11, 21, and 31 after transplantation. Patients received a transfusion of red blood cells if their hemoglobin levels were below 70 g/L, or transfusion of platelets if their platelet levels dropped below 20×109/L. G-CSF (5 µg/kg per day s.c.) was given to all recipients from day 6 after transplantation until their neutrophil count reached 0.5×109/L for three consecutive days. All blood products were irradiated at 2500 cGy before infusion.
Acyclovir was given orally from days −10 to +30 and ganciclovir 5 mg/kg twice daily was routinely administrated intravenously from days −10 to −2. Patient blood was monitored weekly for cytomegalovirus (CMV) DNA by real-time PCR or with a CMV pp65 antigenemia test, and CMV-positive patients were treated with either ganciclovir or foscarnet. CMV-related idiopathic interstitial pneumonia (IP) was defined according to reported criteria. Surveillance for bacterial, fungal, Pneumocystis carinii, and other viral infections was performed based on clinical requirements.
Definition and statistics
The incidence and severity of GVHD was defined by the criteria described by Glucksberg et al. Citation19. The time to GVHD was defined as the time between the day of allo-HSCT and the day that onset of any grade of GVHD was detected.
Patients were evaluated for acute GVHD after successful engraftment was determined, and for chronic GVHD if they survived for at least 100 days after HSCT. Overall survival (OS) and leukemia-free survival (LFS) were evaluated using the Kaplan-Meier analysis. Distributions for time-to-acute/chronic GVHD, time-to-relapse were analyzed with cumulative incidence test, which accounts for the competing risk. All variables were measured from the date of HSCT (day 0). Values were adjusted for engraftment at time of death if the patient did not engraft; for GVHD, survival, and relapse at last follow-up. The end point of the last follow-up for all surviving patients was 31 December 2006. Unless otherwise specified, all reported P-values were based on two-sided hypothesis tests. Alpha was set at 0.05. A statistical software package (SPSS 13.0) was used for all analyses.
Results
Characteristics of patients and grafts
Characteristics of the patients are summarized in . Patients in groups 1 and 2 were comparable, except for donor-recipient gender match. Three patients received G-CSF-primed bone marrow (G-BM), 1 received G-CSF-mobilized peripheral blood stem cell grafts (G-PB), 85 received mixture grafts of G-BM and G-PB, and 4 received mixture grafts of G-BM and G-PB plus umbilical cord blood. The number of hematopoietic mononucleated cells (MNC) was 7.23 (range 4.04–14.12)×108/kg, and CD34+ and CD3+ cells were approximately 2.27 (range 0.34–8.33)×106/kg and 1.65 (range 0.22–9.43)×108/kg, respectively.
Engraftment
All patients achieved hematopoietic recovery after transplantation. The median time for myeloid engraftment was 14 days (range 10–24 days) and for platelet 17 days (range 9–151 days). One patient had secondary graft failure on day 240 after transplantation. She received a second allo-HSCT from the original donor (father), achieved full donor hematological reconstitution, and is still in leukemia-free survival 19 months after transplantation now. One patient received allo-HSCT as a salvage therapy due to graft failure after cord blood transplantation (CBT).
There was no statistically significant association between the extent of HLA disparity and the time of engraftment.
Acute GVHD
Among the 93 patients, 33 (35.5%) had no acute GVHD, 26 (27.96%) had grade I, 16 (17.20%) had grade II, 7 (7.53%) had grade III, and 11 (11.83%) had grade IV. At 100 days after transplantation, the cumulative incidence of acute GVHD was 64.52% (CI 55.49%–75.01%) and grade III–IV was 26.45% (CI 17.51%–39.95%). Factors that might influence the incidence of grade III–IV GVHD were analyzed, which included age and gender of the patients and donors, number of MNC and CD3 cells infused at transplantation, stage of disease before transplantation, and HLA disparity ( and , and III). There was no risk factor except for HLA-B plus -DR mismatches between the recipient-donor pairs (RR 11.208, 95% CI 1.454–86.402, P=0.020). The cumulative incidence of II–IV and III–IV acute GVHD in patients with HLA-B plus HLA-DR mismatched donors was significantly higher than in those without (P=0.011 and 0.015, respectively) after haploidentical transplantation (). Grade II–IV acute GVHD occurred in 5 of 16 patients with HLA mismatched in one locus, and in 29 of 77 patients mismatched in two and three loci; the lack of significance is probably due to the small numbers in group 1 ().
Figure 1. Cumulative incidence of II–IV and III–IV acute graft-versus-host disease (GVHD) after haploidentical transplantation. A: Comparison of the incidence of II–IV acute GVHD between patients with human leukocyte antigen (HLA)-B plus HLA-DR mismatched donors and those without (P=0.011); B: Comparison of the incidence of III–IV acute GVHD between patients with HLA-B plus HLA-DR mismatched donors and those without (P=0.015).
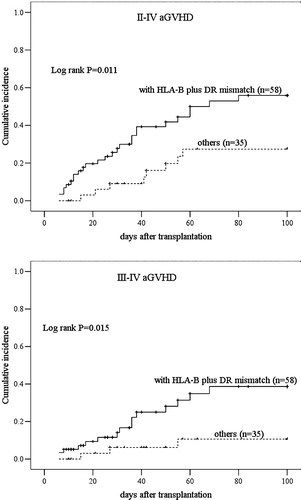
Table III. Clinical outcome in relation to the number of HLA mismatches.
Acute GVHD of grade II or higher was treated with methylprednisolone (0.5–1 mg/kg per day). GVHD manifesting in the skin was treated with methotrexate Citation20. When there was inadequate or absence of response to primary therapy, anti-Tac monoclonal antibody (Daclizumab; Roche, Basel, Switzerland) was administered at 1 mg/kg i.v. on days 1, 3, 8, then at intervals of 7 days for a total of 3–6 doses. Of the 11 patients who developed grade IV acute GVHD, 6 died, one of which died of refractory GVHD, and the remaining 5 died of subsequent infection post treatment. Of the surviving five patients, two remained free of chronic GVHD through the last follow-up, two patients have extensive chronic GVHD, and one has local chronic GVHD. Of the seven patients who suffered from grade III acute GVHD, two died of GVHD, and the other five recovered and survived through the end of study, three of whom have local chronic GVHD. There was no statistically significant correlation between the extent of HLA disparity and the incidence of acute GVHD ( and ).
Chronic GVHD
Among the 81 patients who survived over 100 days after transplantation, 40 developed chronic GVHD. Of these, 22 (55%) had limited chronic GVHD, and 18 (45%) had extensive chronic GVHD. The cumulative incidence of total chronic GVHD was 61.79% (CI 49.33%–77.39%), while 28.93% (CI 19.25%–43.46%) had extensive chronic GVHD at 2 years after transplantation. Chronic GVHD was diagnosed in 8 of 16 patients with HLA mismatched in one locus, 12 of 28 patients mismatched in two loci, and 20 of 37 patients mismatched in three loci. The development of chronic GVHD was not associated with the age or gender of the patients and donors, the extent of HLA disparity, or the stage of disease before transplantation. There was no difference in the incidence of extensive chronic GVHD in patients with different numbers of MNC, CD4, CD8, and CD34 cells, nor in different age groups. The incidence of extensive chronic GVHD in patients who received more CD3 is higher (≥1.66×108/kg) (P=0.035).
There was no statistically significant association between the extent of HLA disparity and the incidence of chronic GVHD.
Relapse, DLI, and imatinib
Nine of all 93 patients relapsed after transplantation, and 5 of them were from the BC group. The incidences of relapse in CML-CP1, CP2, AP, and BC groups pre-HSCT were 3.77%, 0%, 13.94%, and 38.46% respectively, and the difference was determined to be statistically significant (P = 0.0001). The incidences of 4-year relapse in CML-CP and non-CP groups pre-HSCT were 3.29% (CI 0.48%–22.53%) and 31.45% (CI 17.37%–56.96%), respectively, and the difference was determined to be statistically significant (P=0.003).
Among the 93 patients, 4 received a prophylactic DLI. DLIs were delivered at: days 39, 96, 120, and 88, respectively. Acute GVHD grades II–III occurred in three patients that survived HSCT free of their original leukemia. One patient relapsed 10 days after prophylactic DLI and died after further 80 days. Therapeutic DLI with or without imatinib myeslate (Gleevic) was performed in nine relapsed patients, including one patient who relapsed after receiving DLI for prophylaxis. The nine patients were in varying stages of disease prior to transplantation: five were in BC, two in AP, and two in CP. At the time of relapse, five were in hematological relapse state, three in cytogenetic relapse, and one was in a cytogenetic relapse state and also had central system leukemia. Two cases receiving HSCT in BC showed no response to DLI therapy and died of relapse, one case receiving HSCT in AP died of DLI-related GVHD, one case died of severe infection after revolving cytogenetic relapse by DLI, and one case got molecular response and is still in leukemia-free survival through the end point of follow-up. The patient with central system leukemia in cytogenetic relapse achieved continual complete remission after treatment by imatinib mesylate. Of the remaining three patients, one case died of DLI-related GVHD, and two died of relapse ().
Table IV. Characteristics of relapsed patients and outcomes.
Opportunistic infections
Seventy-five episodes of opportunistic infections occurred in 61 of 93 patients within the duration of follow-up. The median time for opportunistic infection was 60 days (range: 0–1056 days) after transplantation. The infected loci included lung (36 episodes), skin (3 episodes), bladder (13 episodes), gastrointestinal tract (8 episodes), central nervous system (2 episodes), and so on. The infections on skin were caused by varicella zoster virus (2 episodes) or Aspergillus species (1 episode). The pneumonia-related pathogens were bacteria (4 episodes), Aspergillus (6 episodes), mold fungus (1 episode), atypical mycobacteria (1 episode), cytomegalovirus (5 cases), and no cases of Pneumocystis carinii. Three episodes were negative in pathogen detection and exhibited a response for glucocorticoids, and two were diagnosed as idiopathic pneumonia syndrome (IPS) and one diagnosed as bronchitis obliterans. No pathogenic evidence was found in the remaining 15 cases, which were deemed as mixture of pneumonia and treated with combination of antifungals/antibiotics. The cumulative incidence of opportunistic infections at 2 years after transplantation was 68.5%.
Survival and follow-up
Up until 31 December 2006, 70 patients were alive and free of their original leukemia through follow-up with a median of 728 days (range 66–1655 days). A total of 23 out of the 93 patients died, comprised of 14 patients in CP or CP2, and 9 patients in AP or BC. Six of the 23 cases died from recurrent leukemia, and 17 from transplant-related complications. The causes of transplantation-related death included GVHD (3 cases), infection (6 cases), interstitial pneumonia or IP (6 cases), and other causes such as heart failure and hepatic failure (2 cases). The non-relapse mortality was 8.72% for 100 days, 20.72% for 1 year, and 20.72% for 2 years. The 100-d transplantation-related mortality (TRM) of patients in CP1, CP2/CR2, AP, and BC are 7.8%, 7.1%, 13.3%, and 7.7%, respectively. The 1-year TRM of patients in CP1, CP2/CR2, AP, and BC are 28.3%, 16.92%, 13.33%, and 7.69%, respectively. No statistically significant difference was identified among them (P=0.622).
The probability of 1-year and 4-year LFS was 76.5% and 74.5% for CP1 patients, 85.7% and 85.7% for CP2/CR2 patients, 80% and 66.7% for AP patients, and 53.8% and 53.8% for BC patients (P=0.221). At present, two out of nine patients who relapsed after transplantation achieved complete molecular response (CMR) and survived free of leukemia.
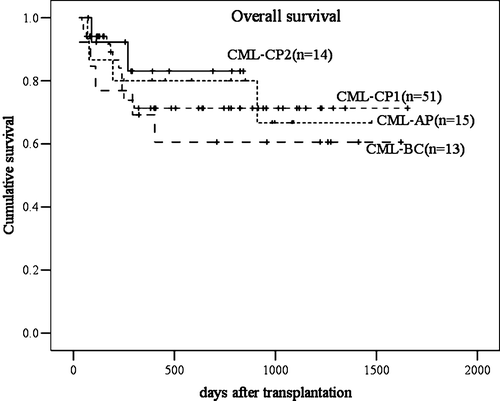
The probability of 4-year OS was 76.5% for CP1 patients, 85.7% for CP2/CR2 patients, 73.3% for AP patients, and 61.5% for BC patients (P=0.7442, ).
Figure 2. Comparison of overall survival in four groups of patients defined according to the stage of disease (chronic phase (CP) 1, CP2, acute phase (AP), blast crisis (BC)). The best is patients transplanted in CP2, then patients in CP1, AP, and BC. No significant differences were found in our transplant protocol.
Overall survival was not predicted by the European Group for Blood and Marrow Transplantation (EBMT) score (). Patients within score ranges of 1 to 2, 3 to 4, and greater than 4 had a similar overall survival (P=0.713). The outcome of the 93 patients divided according to the degree of HLA-disparity between recipient and donor as group 1 and group 2 were comparable, as among patients with HLA class I (including A, B, and A + B alleles), class II (including DR allele), or class I + II (including A + DR, B + DR, and A + B+DR) () mismatched donors ( and ). Multivariate analysis showed that factors affecting transplantation outcomes were HLA-B + DR mismatches versus others for II–III acute GVHD and III–IV acute GVHD and the stage of disease at transplantation for relapse as well as the time from diagnosis to transplant for LFS, OS, and TRM ().
Table V. Multivariate analysis: factors affecting transplantation outcome.
Discussion
As for many hematological diseases, the major concerns for HLA-mismatched/haploidentical HSCT are GVHD and graft rejection. Results from EBMT showed that 103 patients with CML were treated by bone marrow transplantation from haploidentical family members, including 57% patients without in vitro T cell depletion, and the OS and LFS at 5 years was 32% and 25%, respectively Citation9. Fifty-nine patients were transplanted during the first CP, and the probability of survival at 2 years was 47%. Forty-four patients received their transplants for advanced disease, and their probability of survival at 2 years was 25%. Patients transplanted in first CP from a donor mismatched for 0–1 HLA antigens had a probability of survival of 52% at 2 years, and 19% survival rate was observed in patients transplanted in advanced disease stage from donors mismatched for 2–3 HLA antigens. The III–IV acute GVHD patients who received unmanipulated marrow had 34% survival, while 28% survived in recipients of T cell-depleted marrow Citation9. Promising results were found after haploidentical transplantation without ex vivo T cell depletion in our protocol, and all the 93 patients achieved complete donor engraftment. Four-year OS was 75.3%, and III–IV acute GVHD was only observed in 19.4% of the patients. As proposed by Lu et al. and Huang et al. Citation14, Citation15, several reasons can contribute to the results. First, an intensified conditioning regimen was used and ATG was included, which could promote the engraftment and reduce the incidence of GVHD. Second, a strong immunosuppression regimen containing CsA + MTX + MMF was used as the prophylaxis of GVHD. Third, G-CSF-primed allogeneic bone marrow cells and G-CSF-mobilized peripheral blood stem cells were used as source of graft, which contain a large amount of functional modified T cells, and can contribute to prompt and stable donor chimerism and a relatively lower incidence of GVHD Citation20, Citation21.
As we discussed previously, there were no differences in cumulative incidence of acute GVHD in patients with one HLA-mismatched locus, two mismatched loci, or three mismatched loci receiving haploidentical transplantation Citation15. However, we found that HLA mismatch in B plus DR loci is a risk factor for grade III–IV acute GVHD in CML patients. Morishima et al. have demonstrated that single disparities of the HLA-A, -B, -C, or -DRB1 allele between recipients and donors in unrelated HSCT are independent risk factors for acute GVHD Citation22. Petersdorf et al. also showed that the presence of HLA-DRB1 allele mismatching between patients with CML and their unrelated donors was independently associated with increased incidence of grades III–IV acute GVHD Citation23. No confirmed observation that HLA mismatch in B plus DR loci has any synergistic effect on grade III–IV acute GVHD was found in CML patients after transplantation. The mechanism underlying this phenomenon is still unclear now and warrants further investigation. Moreover, there is no non-inherited maternal antigens (NIMA) or non-inherited paternal antigens (NIPA) rule Citation13 in our transplant protocol.
We found that the cumulative incidence of opportunistic infections at 2 years after transplantation was 68.5%. The relatively high infection rates observed in our transplant protocol may be related to the immunological therapy for the severe acute or extensive chronic GVHD. Moreover, immune reconstitution after haploidentical transplantation could also be correlated with the high infection rates, which is currently being investigated in our institute.
It is generally accepted that the outcome of HLA-matched sibling transplants for CML patients in accelerated phase is worse than for those in chronic phase. In a Seattle analysis of 58 patients with accelerated phase CML transplanted from an HLA-identical sibling, the 4-year probabilities of survival and event-free survival were 49% and 43%, respectively Citation24. Even in chronic phase, early transplantation will achieve better results. Data from 4267 recipients of matched-sibling transplants reported to the International Bone Marrow Transplant Registry (IBMTR) between 1994 and 1999 show a probability of survival of 69±2% for 2867 patients transplanted within the first year from diagnosis, and 57±3% for 1391 patients transplanted more than 1 year from diagnosis Citation25. In our study, however, LFS for CML patients in AP was similar to patients in CP, and no difference was observed in the incidence of relapse for patients in CP1 and in AP or CP2. This result may indicate that a strong GVL effect may occur in HLA-mismatched/haploidentical transplantation. The effect of DLI cannot be excluded completely.
In virtually all studies, the outcome of transplantation for patients in blast phase is very poor, due to both a high risk of disease recurrence and a high incidence of transplant-related deaths. The Seattle team had performed transplants for 66 patients with accelerated-phase CML, and the projected 5-year survival rate was estimated at 18%. The actuarial relapse risk 2.5 years post transplant was 100% in patients administered T cell-depleted marrow as compared with 25% in patients administered unmodified marrow Citation26. In our study, even for patients in BC, 4-year LFS can reach levels of 61.5%, much better than those in HLA-matched sibling donor transplantation. Relatively lower TRM may be the main cause of the positive result in BP patients, which may be associated with Gleevic usage pre-HSCT Citation2, Citation27. Prior to the development of imatinib, only a small proportion of patients with blast phase CML could achieve a hematologic remission when treated with chemotherapy. Response rates of patients with CML in blast phase to imatinib are considerably higher than seen with conventional chemotherapy Citation1, Citation2, Citation27, Citation28. It is therefore conceivable that the strong GVL effect after haploidentical HSCT, application of imatinib, and DLI may contribute to the improved outcomes of CML patients in BC.
Novel agents for the treatment of CML have and will continue to emerge, offering patients the opportunity for substantial benefit Citation29. However, when considering currently available treatment options, patients and physicians must recognize that only transplantation has clearly been shown to cure CML, particularly for patients in AP and BP. Our preliminary data for CML patients indicate that in the era of Gleevic transplantation, even from an HLA-mismatched/haploidentical family donor, can be considered as a therapeutic choice for CML patients in AP or CP2 because similar disease free survival (DFS) has been achieved as in patients in CP1. Although no statistically significant differences were found, the outcome of patients in BC was somewhat poorer than patients in CP and AP. It may be a reasonable approach to postpone the HLA-mismatched transplantation for CML until unstable signs of disease progression occurred.
In conclusion, considering the excellent results of tyrosine kinase inhibitor therapy, the CML patient in AP or BC should be a candidate for haploidentical transplantation if there is no HLA-identical sibling donor. However, early transplantation (time from diagnosis to transplant ≤450 d) would achieve better results. The optimal time for HSCT in CML patients from HLA-mismatched family donors needs to be further studied.
Contributions of each author
XJ Huang: Principal Investigator, designed and wrote study; wrote the manuscript.
LP Xu: Study design discussions, treated patients at Peking University Institute of Hematology.
KY Liu: Study design discussions, treated patients at Peking University Institute of Hematology.
All others: Study design discussion, critical analysis of results and manuscript.
Acknowledgements
This work is supported by New Century Excellent Talents in University (NECT-04-0011), Scientific Research Fund for Capital Medicine Development (grant no. 2006-1010), HI-tech research development program of China (grant no. 2006AA02Z4A0) and Program for Innovative Research Team in University (IRT0702). We would like to thank San Francisco Edit (www.sefedit.net) for their assistance in editing this manuscript.
References
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005; 105: 2640–53
- Grigg A, Hughes T. Role of allogeneic stem cell transplantation for adult chronic myeloid leukemia in the imatinib era. Biol Blood Marrow Transplant. 2006; 12: 795–807
- Passweg JR, Walker I, Scbocinski KA, Klein JP, Horowitz MM, Giralt SA. Validation and extension of the EBMT Risk Score for patients with chronic myeloid leukemia receiving allogeneic haematopoietic stem cell transplants. Br J Haematol. 2004; 125: 613–20
- Baron F, Maris MB, Storer BE, Sandmaier BM, Stuart MJ, Mcsweeney PA, et al. HLA matched unrelated donor hematopoietic cell transplantation after nonmyeloablative conditioning for patients with chronic myeloid leukemia. Biol Blood Marrow Transplant. 2005; 11: 272–9
- Carreras E, Jiménez M, Gómez-García V, de la Cámara R, Martín C, Martínez F, et al. Donor age and degree of HLA matching have a major impact on the outcome of unrelated donor haematopoietic cell transplantation for chronic myeloid leukaemia. Bone Marrow Transplant. 2006; 37: 33–40
- Benito AI, Diaz MA, Gonzalez-Vicent M, Sevilla J, Madero L. Hematopoietic stem cell transplantation using umbilical cord blood progenitors: review of current clinical results. Bone Marrow Transplant. 2004; 33: 675–90
- Gluckman E, Rocha V, Arcese W, Michel G, Sanz G, Chan KW, et al. Factors associated with outcomes of unrelated cord blood transplant: guidelines for donor choice. Exp Hematol. 2004; 32: 397–407
- Drobyski WR, Klein J, Flomenberg N, Pietryga D, Vesole DH, Margolis DA, et al. Superior survival associated with transplantation of matched unrelated versus one-antigen disparate haploidentical family donor marrow grafts for the treatment of hematologic malignancies: establishing a treatment algorithm for recipients of alternative donor grafts. Blood. 2002; 99: 806–14
- Speiser DE, Hermans J, Van Biezen A, Starobinski M, Jeannet M, Jacobsen N, et al. Haploidentical family member transplant for patients with chronic myeloid leukemia: a report of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1997; 19: 1197–203
- Ottinger HD, Ferencik S, Beelen DW, Lindemann M, Peceny R, Elmaagacli AH, et al. Hematopoietic stem cell transplantation: contrasting the outcome of transplantations from HLA-identical siblings, partially HLA-mismatched related donors, and HLA-matched unrelated donors. Blood. 2003; 102: 1131–7
- Aversa F, Terenzi A, Felicini R, Carotti A, Falcinelli F, Tabilio A, et al. Haploidentical stem cell transplantation for acute leukemia. Int J Hematol 2002; 7(Suppl)165–8
- Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. 1998; 339: 1186–93
- Van Rood JJ, Loberiz FR, Jr, Zhang MJ, Oudshoorn M, Claas F, Cairo MF, et al. Effect of tolerance to noninherited maternal antigens on the occurrence of graft-versus-host disease after bone marrow transplantation from a parent or an HLA-haploidentical sibling. Blood. 2002; 99: 1572–7
- Lu DP, Dong LJ, Wu T, Huang XJ, Zhang MJ, Han W, et al. Conditioning including antithymocyte HLA-mismatched/haploidentical blood and marrow transplantation can achieve comparable outcomes with HLA-identical sibling transplantation. Blood. 2006; 107: 3065–73
- Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W, et al. Haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion for the treatment of hematological disease. Bone Marrow Transplant. 2006; 38: 291–7
- Huang XJ, Chang YJ, Zhao XY. In vivo induction of T-cell hyporesponsiveness and alteration of immunological cells of bone marrow grafts using granulocyte colony-stimulating factor. Haematologica. 2004; 89: 1517–24
- Liang X, Huang XJ. The differences between recombinant human granulocyte colony-stimulating factor-primed and non-primed peripheral blood stem cell harvests. Chin J Intern Med. 2007; 46: 566–8
- Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W. Donor lymphocyte infusion for the treatment of leukemia relapse after HLA-mismatched/habloidentical T-cell-replete hematopoietic stem cell transplantation. Haematologica. 2007; 92: 414–7
- Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HLA-matched sibling donors. Transplantation. 1974; 18: 295–304
- Huang XJ, Jiang Q, Chen H, Xu L, Liu D, Chen Y, et al. Low-dose methotrexate for the treatment of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2005; 36: 343–8
- Huang XJ, Chang YJ, Zhao XY. Maintaining hyporeponsiveness and polarization potential of T cells after in vitro mixture of G-CSF mobilized peripheral blood grafts and G-CSF primed bone marrow grafts in different proportions. Transplant Immunol. 2007; 17: 193–7
- Morishima Y, Sasazuki T, Inoko H, Juji T, Akaza T, Yamamoto K, et al. The clinical significance of human leukocyte antigen (HLA) allele compatibility in patients receiving a marrow transplant from serologically HLA-A, HLA-B, and HLA-DR matched unrelated donors. Blood. 2002; 99: 4200–6
- Petersdorf EW, Kollman C, Hurley CK, Dupont B, Nademanee A, Begovich AB, et al. Effect of HLA class II gene disparity on clinical outcome in unrelated donor hematopoietic cell transplantation for chronic myeloid leukemia: the US National Marrow Donor Program Experience. Blood. 2001; 98: 2922–9
- Clift RA, Buckner CD, Thomas ED, Bryant E, Anasetti C, Bensinger WI, et al. Marrow transplantation for patients in accelerated phase of chronic myeloid leukemia. Blood. 1994; 84: 4368–73
- International Bone Marrow Transplantation Registry. Available at: http://www.ibmtr.org.2002.
- Martin PJ, Clift RA, Fisher LD, Hansen JA, Appelbaum FR, Doney KC, et al. HLA-identical marrow transplantation during accelerated-phase chronic myelogenous leukemia: analysis of survival and remission duration. Blood. 1988; 72: 1978–84
- Olavarria E, Craddock C, Dazzi F, Marin D, Marktel S, Apperley JF, et al. Imatinib mesylate (STI571) in the treatment of relapse of chronic myeloid leukemia after allogeneic stem cell transplantation. Blood. 2002; 99: 3861–2
- Oehler VG, Gooley T, Synder DS, Johnston L, Lin A, Cummings CC, et al. The effects of imatinib mesylate treatment before allogeneic transplant for chronic myeloid leukemia. Blood. 2007; 109: 1782–9
- Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, et al. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci USA. 2005; 102: 1992–7