Abstract
Genetic defects in glycosyltransferases are responsible for a number of developmental defects and diseases known as congenital disorders of glycosylation (CDGs). Peters’-plus syndrome, a rare autosomal recessive disorder, is now known to be a CDG. This syndrome is characterized by a specific malformation of the eye that includes corneal opaqueness and iridocorneal adhesions (Peters’ anomaly). Affected individuals are short in stature and have short limbs, and may have cleft lip/palate, defects in the central nervous system, heart, and various other organs. The phenotype varies in severity, ranging from death in early childhood to a general delay in growth and development, and is often associated with mental retardation. The mutations responsible for Peters’-plus syndrome inactivate a β1,3-glucosyltransferase whose function is to add a glucose moiety to O-linked fucose, forming a rare glucose-β1,3-fucose disaccharide. This disaccharide modification is specific to thrombospondin type 1 repeats (TSRs), domains found in extracellular proteins that function in cell-cell and cell-matrix interactions and signalling. Some ninety human proteins contain TSRs, but thus far the disaccharide has been demonstrated on only thrombospondin 1, properdin, F-spondin, ADAMTS-13, and ADAMTSL-1. These proteins perform essential functions in embryonic development, tissue remodelling, angiogenesis, neurogenesis, and complement activation. Identification of the β1,3-glucosyltransferase and its substrate proteins is a key step towards understanding their roles in human development, and to uncovering the molecular and cellular mechanisms underlying the clinical manifestations of Peters’-plus syndrome.
Introduction
Glycosylation, the posttranslational addition of oligosaccharides, is a common modification in secreted and cell surface proteins. Oligosaccharides are structurally diverse and rich in chemical information, and therefore ideally suited for cellular communication Citation1. These properties have been amply exploited by nature, and various cell surface glycoproteins and glycolipids are important mediators of cellular recognition, adhesion, and signalling. As the oligosaccharide moiety (also called glycan) is often crucial for the biological activity of the macromolecule, these structures play indispensable roles during embryonic development and in mature tissues Citation2, Citation3. In addition, cell surface glycans are intimately involved in the pathogenesis of many diseases, including cancer Citation4, Citation5.
Glycan biosynthesis proceeds by the sequential addition of single sugar units in pathways catalysed by glycosyltransferases Citation6, Citation7. These enzymes are located in the endoplasmic reticulum (ER) and the Golgi apparatus, where they act on the substrate glycoproteins en route to the cell surface. The total number of human glycosyltransferases is well over 200, as indicated by the known or potential enzyme sequences listed in a database of carbohydrate-active enzymes Citation8. A given cell or tissue expresses only a subset of the total enzyme complement, synthesizing the glycan profile specific to that cell or tissue.
Genetic defects in glycosyltransferases and other glycan-processing enzymes are responsible for a number of diseases, collectively known as congenital disorders of glycosylation (CDGs). To date, 28 such diseases have been described, and the list is still growing Citation9–11. This list is likely to eventually include several forms of muscular dystrophy Citation12. The various glycosylation defects have diverse phenotypic manifestations, which can be severe, underscoring the biological importance of glycosylation in human development.
Abbreviations
Key messages
Peters’-plus syndrome is an autosomal recessive disorder that comprises developmental defects of the eye, brain, skeleton, and other organs. It is now known to be a congenital disorder of glycosylation caused by a defect in a β1,3-glucosyltransferase (b3Glc-T).
The function of the glucosyltransferase is to complete the synthesis of an O-linked glucose-fucose modification on thrombospondin type 1 repeats, domains found in extracellular signalling proteins that function in angiogenesis, neuronal guidance, and tissue remodelling during development.
Identification of the glucosyltransferase and its substrate proteins is a key step towards understanding the molecular and cellular mechanisms underlying the developmental defects observed in this syndrome.
Peters’-plus syndrome
In 1906, Peters described a malformation of the eye that came to be known as Peters’ anomaly Citation13. Its most diagnostic features are corneal opacity (leukoma), variable iridolenticular attachments to the cornea, absent or defective Descemet membrane, and thinning of the posterior aspect of the cornea. Other conditions that are often present are glaucoma and cataracts. Peters’ anomaly is a major developmental defect in the anterior chamber of the eye, which may result from disrupted function of neural crest cells early in embryogenesis Citation14.
Further study of afflicted individuals revealed that Peters’ anomaly is often associated with a host of other, extraocular developmental defects, and this expanded set of symptoms was named Peters’-plus syndrome by van Schooneveld et al. in 1984 Citation15. Some of the major clinical findings in this syndrome, also known as Kivlin-Krause syndrome, are summarized in and reviewed in reference Citation14. A more detailed clinical synopsis as well as more information about the syndrome can be found at the Online Mendelian Inheritance in Man (OMIM) web site Citation16. The syndrome follows an autosomal recessive mode of inheritance and is therefore uncommon. The medical literature contains reports of fewer than 70 affected individuals, but the true prevalence of the syndrome is unknown.
Table I. Common defects in Peters’-plus syndrome, and their frequencies (%).
Peters’-plus syndrome is associated with characteristic craniofacial features, which include a prominent forehead, ocular hypertelorism, narrow palpebral fissures, a thin ‘cupid's bow’ upper lip, and long philtrum. A cleft lip is common, and can be accompanied by a cleft palate. The symptoms are dramatically evident in the central nervous system, as virtually all patients show some degree of mental retardation, which can be severe and accompanied by cortical underdevelopment or atrophy. The skeletal system shows obvious signs of delayed or abnormal development, manifested as short stature and short limbs (short-limb dwarfism) as well as broad hands. Overall, the phenotype shows considerable variation, but the total spectrum of symptoms is vast, covering virtually all major internal organs, including the heart, kidney, and reproductive system.
The genetic defect in Peters’-plus syndrome
The genetic defect responsible for Peters’-plus syndrome was identified in 2006 by Lesnik Oberstein et al. Citation17, who were led to a certain region of chromosome 13 after detecting a microdeletion there by array-based comparative genomic hybridization. While the microdeletion was only found in two afflicted brothers and covered a number of loci, sequence analysis of DNA from more patients pin-pointed the mutated locus. It is a previously identified and characterized gene Citation18 that was originally discovered in a cell culture system used to model differentiation of intestinal epithelial cells Citation19–21. The gene resides in chromosome 13 (at 13q12.3) where it spans a length of 132,188 bp, and its coding region is divided into 15 exons by 14 introns. The gene product was predicted to be a novel glycosyltransferase with a clearly identifiable catalytic domain and other structural features typically found in these enzymes Citation18.
The mutation that causes Peters’-plus syndrome does not reside in the coding region of the gene but in an intron, where the first nucleotide of intron 8 is an A instead of the wild-type G (). This point mutation disrupts the donor splice site and causes defective splicing of the pre-mRNA, from which exon 8 is erroneously spliced out. As a result, the reading frame in the mature mRNA is disrupted, translation stops prematurely and the protein product lacks the catalytic domain. In a sample of 20 Peters’-plus patients, all were found to carry this same mutation Citation17; 16 patients were homozygous and the 2 brothers carrying the microdeletion were hemizygous for the mutation. The remaining two patients, who were siblings, carried this mutation in only one (maternal) allele, but they were found to carry a second mutation in the paternal allele of the same gene. This second mutation is also in an intron (the fifth nucleotide in intron 5 is A instead of G) (), and it also disrupts intron splicing, causing a truncated translation product without a catalytic domain.
Figure 1. The b3Glc-T gene. The exons are shown as rectangles:□=5′- and 3′-UTRs;▪ = signal peptide;□ = the ‘stem’ domain;□ = the catalytic domain. The introns are denoted by discontinuous lines. The two mutations found in Peters’-plus patients are indicated by the vertical arrows, and their locations relative to cDNA co-ordinates (reference 17) are given in the rectangles above. The horizontal bar indicates the scale for the exons; the exact lengths of the exons and introns can be found in reference Citation18.
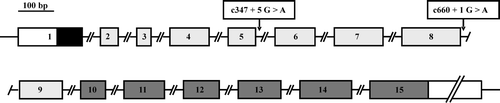
The analysis by Lesnik Oberstein et al. Citation17 demonstrated that Peters’-plus syndrome is monogenic, and identified the genetic locus involved. The syndrome is primarily caused by a single mutation, found in all patients studied, although two siblings also had a second mutation in one allele. This analysis provides a potential molecular diagnostic tool for Peters’-plus syndrome, although it remains to be seen if the syndrome is genetically as homogeneous as this first, limited sample of 20 patients seems to indicate.
The gene in question encodes a 498-amino-acid protein that displays an overall architecture and primary structural features that identify it as a glycosyltransferase Citation18. In its N-terminus the protein has a signal peptide (residues 1–28) that directs it to the ER. This region was first erroneously identified as a transmembrane domain due to its helical and hydrophobic nature but, as pointed out by Sato et al. Citation22 and Kozma et al. Citation23, it is in fact an ER signal peptide. The signal peptide is followed by a larger domain (residues 29–260) that was originally called the ‘stem region’ by analogy to other, Golgi apparatus-resident glycosyltransferases. This domain, however, appears substantially more structured than conventional glycosyltransferase stems and has features that indicate it may be involved in carbohydrate binding or substrate glycoprotein recognition Citation18. The C-terminal portion of the protein (residues 261–498) is a clearly identifiable catalytic domain which contains a triplet of aspartate residues (DDD) in its core. This triplet corresponds to the more general DXD motif that is characteristic of enzymes that assume the GT-A structural fold Citation7, where it forms the part of the catalytic site that is involved in binding the donor substrate.
The catalytic domain of the protein shares overall sequence similarity and certain conserved residues and motifs found in β3-glycosyltransferases, enzymes that join two sugars by β1,3-glycosidic linkage. Consequently, the putative enzyme was named β3-glycosyltransferase-like (B3GTL) Citation18. It turns out that the prediction about the enzyme's activity implicit in this name was in fact correct, attesting to the power of bioinformatics and the recent advances in our understanding of glycosyltransferases. The gene was later named β3-galactosyltransferase-like (B3GALTL) in accordance with nomenclature standards of the Human Genome Organization (HUGO).
The B3GTL gene is expressed in a wide range of normal tissues, which display considerable differences in the levels of the mRNA Citation18, Citation22, indicating that the gene is subject to strong tissue-specific regulation. The wide expression profile is consistent with the multitude of tissues known to be affected in Peters’-plus syndrome (see ). In addition to serving a function in those tissues, B3GTL seems to be involved in thyroid development and function, and an association between hypothyroidism and Peters’-plus syndrome has recently been reported Citation24. Interestingly, this gene also plays some role in malignancy, as its expression has been found to be upregulated in thyroid oncocytic tumours Citation25.
It is likely that the B3GTL enzyme is not only important in human development, but performs an essential function in all multicellular animals, as potential orthologues to it are found across the Metazoa, from mammals to insects and nematodes Citation18, Citation26. B3GTL orthologues from various species share certain features, which are particularly obvious in the catalytic domain, the stem domain being more variable. The catalytic domains share approximately 40% sequence identity and a conserved pattern of cysteine residues, which suggests they may assume a similar conformation. Another distinguishing feature of B3GTL orthologues is the presence of a C-terminal motif resembling the tetrapeptide KDEL (the standard one-letter code for amino acids), which is a signal in proteins for retention in the ER compartment of the secretory pathway.
B3GTL is a β1,3-glucosyltransferase
The enzymatic activity of B3GTL was solved in 2006, providing a break-through in understanding its biological function. Two groups, Narimatsu's Citation22 and Hofsteenge's Citation23, demonstrated that the gene product is indeed a β1,3-glycosyltransferase, an enzyme that joins a glucose (Glc) moiety to O-linked fucose (Fuc). The enzyme was found to use uridine 5′-diphosphate (UDP)-glucose as the donor substrate, thus its full name is ‘UDP-glucose: fucose β1,3-glucosyltransferase’ (abbreviated b3Glc-T). The enzyme activity had been detected in Chinese hamster ovary cells in 1999 Citation27, but the first hint of the existence of such an enzyme came as early as 1975 when the Glc-Fuc disaccharide product was detected in human urine Citation28. The relevance of this disaccharide to Peters’-plus syndrome was established definitively in 2008 by Hess et al. Citation29, who studied the Glc-Fuc modification in one of its physiological contexts, on the serum glycoprotein properdin (which is discussed in more detail below). These authors demonstrated, using a sensitive mass spectometric assay and properdin as a ‘reporter protein’, that the Glc moiety is indeed lacking in patients who carry biallelic truncating mutations in the b3Glc-T gene.
The acceptor substrate for b3Glc-T is a fucose moiety that has been linked to a serine (Ser) or threonine (Thr) residue in a polypeptide by O-glycosidic bond (Fuc-α1-O-Ser/Thr). This first step in the synthesis of the disaccharide is catalysed by protein O-fucosyltransferase-2 (POFUT2) Citation30–32, an enzyme that co-localizes with b3Glc-T to the ER Citation23. Thus these two enzymes form a complete glycosylation pathway, synthesizing the mature Glc-β1,3-Fuc-α1-O disaccharide that is not extended any further. Terminal β1,3-linked glucose is extremely rare in human glycoconjugates, in fact thus far it has been found uniquely in this disaccharide.
O-linked fucose occurs in only one other context, a distinct pathway responsible for the synthesis of a tetrasaccharide on protein domains called epidermal growth factor-like repeats (EGFs) Citation30, Citation31. This pathway is initiated by protein O-fucosyltransferase-1 (POFUT1), which adds fucose specifically to EGFs. The second enzyme, a β1,3-N-acetylglucosaminyltransferase called Fringe Citation33, adds N-acetylglucosamine (GlcNAc) which is then extended further with galactose (Gal) and sialic acid (Sia) to form the mature tetrasaccharide Sia-α2,3-Gal-β1,4-GlcNAc-β1,3-Fuc-α1-O-Ser/Thr. EGFs are found in a number of proteins, including Notch, a transmembrane receptor in a signalling pathway that modulates diverse developmental processes. The activity and ligand binding of Notch are regulated by the tetrasaccharide, ablation of which has dramatic developmental consequences Citation33, Citation34.
The thrombospondin type 1 repeat
The polypeptide substrate for the Glc-Fuc modification is a specific protein domain, the thrombospondin type 1 repeat (TSR) (also known as ‘TSP type-1’ or ‘TSP-1’ domain). This domain derives its name from the blood platelet protein thrombospondin 1, but it is also found in a number of other proteins, which contain variable numbers of the repeat Citation35–37, as discussed below. The various TSRs vary in length from approximately 50 to 60 amino acid residues, and the structure of the domain from human thrombospondin 1 has been determined by X-ray crystallography Citation38. The domain consists of three antiparallel strands (see ), an irregular rippled strand (A) and two regular beta strands (B and C), which assume a compact layered fold held together by internally stacked side chains and three disulfide bridges. One face of the structure contains a positively charged spiral groove that is suggested to be the recognition site that mediates interactions with various ligands Citation38.
Figure 2. The thrombospondin type 1 repeat (TSR). A schematic representation of the secondary structural elements, showing the three antiparallel strands as horizontal rectangles (A, B, and C). The loop between strands A and B contains the sequence motif, indicated by the standard one-letter code for amino acids inside the vertical rectangle, to which the Glc-Fuc disaccharide is attached. Glc = glucose; Fuc = fucose.
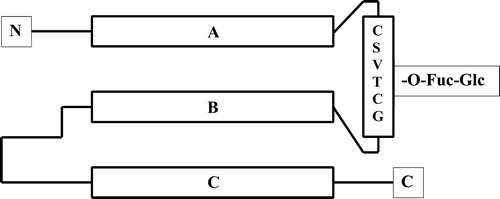
In the TSR fold, the loop between strands A and B contains the sequence motif to which the Glc-Fuc disaccharide is attached (). This motif is CSVTCG (the standard one-letter code for amino acids) in the second and third TSRs of human thrombospondin 1, but variants of this sequence are conserved in other TSRs. A consensus sequence based on human thrombospondin 1 was determined to be CSX(S/T)CG (where X can be any amino acid) Citation39, although a revised consensus (WX5CX2–3S/TCX2G) was suggested after examination of F-spondin and properdin Citation40. The underlined Thr (or Ser) residue in this motif serves as the site to which the Glc-Fuc disaccharide is attached by O-glycosidic bond. The TSR domain also undergoes a second kind of glycosylation, C-mannosylation, on three of its tryptophan (Trp) residues Citation39, but the function of this unusual modification is not known. Moreover, the relationship and possible interdependence of the two types of glycosylation have not yet been explored comprehensively. It is perhaps noteworthy that one of the mannosylated Trp residues is within ten residues of the Glc-Fuc-linked Thr, and it is possible that this part of the domain derives functional contributions from both glycans.
TSR domains are found in some ninety human proteins, but the Glc-Fuc modification has been demonstrated on only native platelet thrombospondin 1 Citation39 and native properdin Citation29, and on recombinant F-spondin Citation40, ADAMTS-13 Citation41, and ADAMTSL-1 Citation42. Thrombospondin 1 (TSP-1) is a large, 450-kDa glycoprotein that is secreted into the extracellular matrix by several types of cell. It is a homotrimer whose monomer subunits are tethered together near their N-termini, forming a globular central domain from which three long flexible arms extend outwards. Each monomer contains three non-identical TSRs as well as a number of other domains, which are discussed in references Citation35–37. TSP-1 is stored in large amounts in the α-granules of blood platelets, the source from which it was first purified, where its tasks pertain to blood clotting. Upon activation by thrombin, TSP-1 binds to the surfaces of activated platelets, promoting their aggregation, and it is also incorporated into the fibrin network of the clot. Although its name derives from these functions in blood clotting, TSP-1 has proven to be a multifunctional protein that has diverse roles in embryonic development, tissue remodelling, wound healing, angiogenesis, and tumour growth.
The role of TSP-1 in angiogenesis is particularly noteworthy. TSP-1 is a potent inhibitor of vascularization and thereby plays a homeostatic role in the maintenance of blood vessel density in tissues. It inhibits angiogenesis by inducing microvascular endothelial cells to undergo apoptosis in a chain of events that begins with binding of TSP-1 to CD36, a transmembrane receptor on the surface of the endothelial cell. This interaction triggers a signalling cascade which acts through p59fyn (a Src family tyrosine kinase), a caspase-3-like protease, and p38 mitogen-activated protein (MAP) kinase Citation43, Citation44. As a result, the endothelial cell is unable to respond to a wide variety of proangiogenic stimuli and ultimately undergoes apoptosis. The first step in this cascade, binding of TSP-1 to CD36, is mediated by the TSRs, and the CSVTCG motif appears to be important for this interaction Citation45. It is possible that the Glc-Fuc disaccharide modulates this interaction, a prospect that has not yet been explored empirically.
F-spondin, also known as vascular smooth muscle cell growth-promoting factor (VSGP), is a protein involved in differentiation of neurons and growth of vascular smooth muscle cells. The 110-kDa polypeptide containing six C-terminal TSR modules is secreted and found in the extracellular matrix in a number of tissues. During embryonic development, F-spondin is expressed in the nervous system, mainly in the hippocampus and the floor plate, where it is required for patterning of axonal growth Citation46. It promotes and guides the outgrowth of sensory, commissural, and hippocampal axons, while inhibiting the outgrowth of motor axons Citation47–49. In addition to its role in neurogenesis, F-spondin has a second function in vascularization, where it appears to be involved in termination of angiogenesis as well as in stabilization of newly formed vessels by promoting the growth and attachment of smooth muscle cells Citation50.
Properdin is a serum glycoprotein that functions in activation of the alternative pathway of the complement system. Native properdin is an unusual oligomer, consisting of two, three, or four monomer units assembled into a cyclic structure. Each monomer unit is a rod-like 53-kDa polypeptide that contains six TSR modules which have been shown to be involved in the intramolecular interactions related to oligomerization, as well as in ligand binding Citation51. One of properdin's ligands is the complement fragment C3b, binding to which is mediated by a specific TSR Citation52. This interaction promotes the association between activated C3b and factor B, stabilizing the C3b-Bb convertase complex, which is then deposited on a pathogen surface Citation53. Because properdin can bind many ligands at a time, it can form a lattice of complexes, thus specifically upregulating the alternative pathway on a surface and not in the fluid phase.
ADAMTS-13, a 160-kDa plasma glycoprotein that contains eight TSRs Citation54, belongs to the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family of proteinases Citation55. ADAMTS-13 regulates the activity of von Willebrand factor by cleaving it into smaller, less thrombogenic forms, and thereby helps to maintain vascular homeostasis Citation54. ADAMTSL-1, also known as punctin-1, is a 60-kDa matrix glycoprotein that contains four TSRs. It belongs to a group of proteins that resemble the ADAMTS metalloproteinases but lack the catalytic domain and proteinase activity. Little is known about the function of these ‘ADAMTS-like’ molecules, but a role in modulation of the ADAMTS proteinases has been suggested Citation56. In the case of both ADAMTS-13 Citation41 and ADAMTSL-1 Citation42, the disaccharide modification was shown to be required for efficient secretion of the recombinant protein into the extracellular matrix.
In addition to those discussed above, more than eighty human proteins contain TSR modules. Some of these do not contain a relevant version of the CSX(S/T)CG consensus motif, and in particular the S/T residue that is required for O-linked glycosylation, and therefore cannot be modified with the Glc-Fuc disaccharide. Proteins that contain the consensus sequence, however, are potential substrates for this modification (some of these proteins are listed in ). In these proteins, the number of TSR modules ranges from 1 in connective tissue growth factor (CTGF) to 25 in subcommissural organ-specific spondin (SCO-spondin) Citation57. Most of these proteins are secreted into the extracellular matrix, but brain-specific angiogenesis inhibitors 1 and 3 (BAIs 1 and 3), semaphorins 5A and 5B, and UNC-5 (named after its Caenorhabditis elegans homologue, ‘unco-ordinated-5’) are transmembrane proteins whose TSRs are located in the extracellular portions.
Table II. Human proteins containing thrombospondin type 1 repeats that are potentially modified with the Glc-Fuc disaccharide.
Many of the proteins listed in are involved in angiogenesis, as exemplified by BAI 1 Citation58, CTGF Citation59 and TSP-2 Citation37, Citation44. Molecules employed in neuronal development are prominently represented on this list, as SCO-spondin Citation60, semaphorin 5A Citation61, and the netrin receptor UNC-5 Citation62, Citation63 have been shown to function in neuronal guidance and patterning. Interestingly, some of these (e.g. UNC-5) are also utilized as path-finding molecules in vascular patterning Citation64, highlighting their multifunctional nature. Another class of proteins, the ADAMTS matrix proteinases Citation55, and their potential modulators papilin Citation65 and the ADAMTSLs Citation56, are involved in tissue degradation and remodelling.
This brief overview shows that many TSR-containing proteins are employed in tissue remodelling and organogenesis and therefore perform essential roles during embryonic development. Although the functions discovered thus far are concentrated on the vascular and nervous systems, it is likely that many of these molecules, like the prototypical TSP-1, are in fact multifunctional and act in multiple tissues. Several of these molecules have been discovered only recently and are just beginning to be investigated, and future research will undoubtedly uncover new functions within this group of proteins whose realm is communication and signalling in the extracellular matrix.
Concluding remarks
Peters’-plus syndrome is now known to be a congenital disorder of glycosylation caused by mutations that inactivate a β1,3-glucosyltransferase (b3Glc-T). While the severe phenotype of this syndrome, reflecting complete lack of enzyme activity, is rare, it is possible that more common mutations and polymorphisms at this locus give rise to milder phenotypes. Since heterozygous relatives of Peters’-plus patients were observed to have an increased incidence of glaucoma Citation17, it is tempting to speculate that polymorphisms or heterozygosity at the b3Glc-T locus may be involved in the genesis of this more common condition, and possibly other extraocular disorders as well.
Identification of the enzyme defect that causes Peters’-plus syndrome is a key step towards understanding the pathogenesis of this syndrome. The function of b3Glc-T is to complete the synthesis of a rare O-linked Glc-Fuc disaccharide on TSRs, domains found in extracellular signalling proteins that perform important functions in development, including angiogenesis and neurogenesis. These proteins will ultimately provide the keys to understanding the molecular and cellular mechanisms underlying the phenotypic manifestations of Peters’-plus syndrome. More insight into the role of the Glc-Fuc modification should come from a mouse model in which the b3Glc-T gene Citation26 has been disrupted, as ‘knock-out’ mice have proven to be very powerful in exploration of mammalian gene function.
In many of the potential substrate proteins of b3Glc-T, the TSR domains have been shown to be involved in functionally important protein-protein interactions, and in one case the site of interaction has been mapped to the motif that contains the Glc-Fuc disaccharide. This raises the intriguing possibility that the disaccharide may modulate these interactions, a hypothesis that is waiting to be explored. At present, the best evidence for the biological significance of the disaccharide comes from Peters’-plus syndrome itself, whose phenotype bespeaks the importance of this seemingly modest structure in human development.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Gabius H-J, Siebert H-C, Andre S, Jimenez-Barbero J, Rudiger H. Chemical biology of the sugar code. Chembiochem. 2004; 5: 740–64
- Muramatsu T. Essential roles of carbohydrate signals in development, immune response and tissue functions, as revealed by gene targeting. J Biochem. 2000; 127: 171–6
- Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004; 73: 491–537
- Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004; 95: 377–84
- Dube DH, Bertozzi CR. Glycans in cancer and inflammation—potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005; 4: 477–88
- Breton C, Mucha J, Jeanneau C. Structural and functional features of glycosyltransferases. Biochimie. 2001; 83: 713–8
- Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006; 16: 29R–37R
- CAZy—Carbohydrate-active enzymes. Available at: www.cazy.org.
- Jaeken J, Carchon H. Congenital disorders of glycosylation: a booming chapter in pediatrics. Curr Opin Pediatr. 2004; 16: 434–9
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007; 8: 261–78
- Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007; 7: 389–96
- Muntoni F, Brockington M, Torelli S, Brown SC. Defective glycosylation in congenital muscular dystrophies. Curr Opin Neurol. 2004; 17: 205–9
- Peters A. Ueber angeborene defektbildung der Descemetschen membran. Klin Monatsbl Augenheilkd. 1906;44:27–40 and 105–19.
- Wenniger-Prick LJ, Hennekam RC. The Peters’ plus syndrome: a review. Ann Genet. 2002; 45: 97–103
- van Schooneveld MJ, Delleman JW, Beemer FA, Bleeker-Wagemakers EM. Peters’-plus: a new syndrome. Ophthalmic Paediatr Genet. 1984; 4: 141–5
- Online Mendelian Inheritance in Man (OMIM). Available at: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=261540.
- Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, et al. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79:562–6. Erratum in: Am J Hum Genet. 2006;79:985.
- Heinonen TYK, Pasternack L, Lindfors K, Breton C, Gastinel LN, Mäki M, et al. A novel human glycosyltransferase: primary structure and characterization of the gene and transcripts. Biochem Biophys Res Commun. 2003; 309: 166–74
- Halttunen T, Marttinen A, Rantala I, Kainulainen H, Mäki M. Fibroblasts and transforming growth factor β induce organization and differentiation of T84 human epithelial cells. Gastroenterology. 1996; 111: 1252–62
- Lindfors K, Halttunen T, Huotari P, Nupponen N, Vihinen M, Visakorpi T, et al. Identification of novel transcription factor-like gene from human intestinal cells. Biochem Biophys Res Commun. 2000; 276: 660–6
- Lindfors K, Halttunen T, Kainulainen H, Mäki M. Differentially expressed CC3/TIP30 and rab11 along in vivo and in vitro intestinal epithelial cell crypt-villus axis. Life Sci. 2001; 69: 1363–72
- Sato T, Sato M, Kiyohara K, Sogabe M, Shikanai T, Kikuchi N, et al. Molecular cloning and characterization of a novel human β1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology. 2006; 16: 1194–206
- Kozma K, Keusch JJ, Hegemann B, Luthet KB, Klein D, Hess D, et al. Identification and characterization of a β1,3-glucosyltransferase that synthesizes the Glc-β1,3-Fuc di-saccharide on thrombospondin type 1 repeats. J Biol Chem. 2006; 281: 36742–51
- Kosaki R, Kamiishi A, Sugiyama R, Kawai M, Hasegawa T, Kosaki K. Congenital hypothyroidism in Peters plus syndrome. Ophthalmic Genet. 2006; 27: 67–9
- Jacques C, Baris O, Prunier-Mirebeau D, Savagner F, Rodien P, Rohmer V, et al. Two-step differential expression analysis reveals a new set of genes involved in thyroid oncocytic tumors. J Clin Endocrinol Metab. 2005; 90: 2314–20
- Heinonen TYK, Pelto-Huikko M, Pasternack L, Mäki M, Kainulainen H. Murine ortholog of the novel glycosyltransferase, B3GTL: primary structure, characterization of the gene and transcripts, and expression in tissues. DNA Cell Biol. 2006; 25: 465–74
- Moloney DJ, Haltiwanger RS. The O-linked fucose glycosylation pathway: identification and characterization of a uridine diphosphoglucose: fucose-beta-1,3-glucosyltransferase activity from Chinese hamster ovary cells. Glycobiology. 1999; 9: 679–87
- Hallgren P, Lundblad A, Svensson S. A new type of carbohydrate-protein linkage in a glycopeptide from normal human urine. J Biol Chem. 1975; 250: 5312–14
- Hess D, Keusch JJ, Lesnik Oberstein SA, Hennekam RCM, Hofsteenge J. Peters plus syndrome is a new congenital disorder of glycosylation and involves defective O-glycosylation of thrombospondin type 1 repeats. J Biol Chem. 2008; 283: 7354–60
- Shao L, Haltiwanger RS. O-fucose modifications of epidermal growth factor-like repeats and thrombospondin type 1 repeats: unusual modifications in unusual places. Cell Mol Life Sci. 2003; 60: 241–50
- Luo Y, Nita-Lazar A, Haltiwanger RS. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J Biol Chem. 2006; 281: 9385–92
- Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 2006; 281: 9393–99
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, et al. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000; 406: 369–75
- Rampal R, Luther KB, Haltiwanger RS. Notch signalling in normal and disease states: possible therapies related to glycosylation. Curr Mol Med. 2007; 7: 427–45
- Adams JC, Tucker RP. The thrombospondin type 1 repeat (TSR) superfamily: diverse proteins with related roles in neuronal development. Dev Dyn. 2000; 218: 280–99
- Tucker RP. The thrombospondin type 1 repeat superfamily. Int J Biochem Cell Biol. 2004; 36: 969–74
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004; 36: 961–8
- Tan K, Duquette M, Liu JH, Dong Y, Zhang R, Joachimiak A, et al. Crystal structure of the TSP-1 type 1 repeats: a novel fold and its biological implication. J Cell Biol. 2002; 159: 373–82
- Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, et al. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J Biol Chem. 2001; 276: 6485–98
- Gonzalez de Peredo A, Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. C-mannosylation and o-fucosylation of thrombospondin type 1 repeats. Mol Cell Proteomics. 2002; 1: 11–8
- Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-fucosylation is required for ADAMTS13 secretion. J Biol Chem. 2007; 282: 17014–23
- Wang LW, Dlugosz M, Somerville RP, Raed M, Haltiwanger RS, Apte SS. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J Biol Chem. 2007; 282: 17024–31
- Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-mediated inhibition of neovascularization by thrombospondin-1. Nat Med. 2000; 6: 41–8
- Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007; 74: 90–9
- Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997; 138: 707–17
- Klar A, Baldassare M, Jessell TM. F-spondin: a gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell. 1992; 69: 95–110
- Burstyn-Cohen T, Tzarfaty V, Frumkin A, Feinstein Y, Stoeckli E, Klar A. F-spondin is required for accurate pathfinding of commissural axons at the floor plate. Neuron. 1999; 23: 233–46
- Feinstein Y, Klar A. The neuronal class 2 TSR proteins F-spondin and Mindin: a small family with divergent biological activities. Int J Biochem Cell Biol. 2004; 36: 975–80
- Zisman S, Marom K, Avraham O, Rinsky-Halivni L, Gai U, Kligun G, et al. Proteolysis and membrane capture of F-spondin generates combinatorial guidance cues from a single molecule. J Cell Biol. 2007; 178: 1237–49
- Terai Y, Abe M, Miyamoto K, Koike M, Yamasaki M, Ueda M, et al. Vascular smooth muscle cell growth-promoting factor/F-spondin inhibits angiogenesis via the blockade of integrin αvβ3 on vascular endothelial cells. J Cell Physiol. 2001; 188: 394–402
- Perdikoulis MV, Kishore U, Reid KBM. Expression and characterisation of the thrombospondin type 1 repeats of human properdin. Biochim Biophys Acta. 2001; 1548: 265–77
- Higgins JM, Wiedemann H, Timpl R, Reid KB. Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type 1 repeats. Identification of modules important for function. J Immunol. 1995; 155: 5777–85
- Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006; 281: 2128–32
- Soejima K, Nakagaki T. Interplay between ADAMTS13 and von Willebrand factor in inherited and acquired thrombotic microangiopathies. Semin Hematol. 2005; 42: 56–62
- Jones GC, Riley GP. ADAMTS proteinases: a multi-domain, multi-functional family with roles in extracellular matrix turnover and arthritis. Arthritis Res Ther. 2005; 7: 160–9
- Hirohata S, Wang LW, Miyaqi M, Yan L, Seldin MF, Keene DR, et al. Punctin, a novel ADAMTS-like molecule, ADAMTSL-1, in extracellular matrix. J Biol Chem. 2002; 277: 12182–9
- Meiniel O, Meiniel A. The complex multidomain organization of SCO-spondin protein is highly conserved in mammals. Brain Res Rev. 2007; 53: 321–7
- Kaur B, Brat DJ, Devi NS, Van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene. 2005; 24: 3632–42
- Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis. 2002; 5: 153–65
- Gobron S, Creveaux I, Meiniel R, Didier R, Herbert A, Bamdad M, et al. Subcommissural organ/Reissner's fiber complex: characterization of SCO-spondin, a glycoprotein with potent activity on neurite outgrowth. Glia. 2000; 32: 177–91
- Kantor DB, Chivatakarn O, Peer KL, Oster SF, Inatani M, Hansen MJ, et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron. 2004; 44: 961–75
- Li W, Aurandt J, Jürgensen C, Rao Y, Guan KL. FAK and Src kinases are required for netrin-induced tyrosine phosphorylation of UNC-5. J Cell Sci. 2006; 119: 47–55
- Round J, Stein E. Netrin signaling leading to directed growth cone steering. Curr Opin Neurobiol. 2007; 17: 15–21
- Eichmann A, Makinen T, Alitalo K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. 2005; 19: 1013–21
- Fessler JH, Kramerova I, Kramerov A, Chen Y, Fessler LI. Papilin, a novel component of basement membranes, in relation to ADAMTS metalloproteases and ECM development. Int J Biochem Cell Biol. 2004; 36: 1079–84
- Koo BH, Goff CL, Jungers KA, Vasanji A, O'Flaherty J, Weyman CM, et al. ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix Biol. 2007; 26: 431–41
- Hall NG, Klenotic P, Anand-Apte B, Apte SS. ADAMTSL-3/punctin-2, a novel glycoprotein in extracellular matrix related to the ADAMTS family of metalloproteases. Matrix Biol. 2003; 22: 501–10
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007; 450: 430–4
- Rus H, Cudrici C, Niculescu F. The role of the complement system in innate immunity. Immunol Res. 2005; 33: 103–12
- Tong ZY, Brigstock DR. Intrinsic biological activity of the thrombospondin structural homology repeat in connective tissue growth factor. J Endocrinol. 2006; 188: R1–8
- Arnott JA, Nuglozeh E, Rico MC, Arango-Hisijara I, Odgren PR, Safadi FF, et al. Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-beta1-induced extracellular matrix production in osteoblasts. J Cell Physiol. 2007; 210: 843–52
- Xu X, Dong C, Vogel BE. Hemicentins assemble on diverse epithelia in the mouse. J Histochem Cytochem. 2007; 55: 119–26