Abstract
Vascular endothelial growth factor (VEGF), a potent growth factor for endothelial cells and inducer of angiogenesis, is important for endothelial integrity and thus for vascular function. On the other hand, VEGF may enhance the pathophysiologic mechanism of plaque formation and plaque destabilization. In this review we discuss the data available so far for VEGF as angiogenic and/or inflammatory cytokine in the vulnerable atherosclerotic plaque.
Introduction
Cardiovascular disease is one of the most significant causes of morbidity and mortality worldwide. Development of atherosclerosis and the rupture of atherosclerotic plaques play important roles in cardiovascular disease. Atherosclerotic plaque formation and plaque instability involve processes such as inflammation, lipid and protein deposition, angiogenesis, and apoptosis and necrosis of various cells Citation[1–3]. Although a large, non-occlusive plaque can cause myocardial ischemia, the acute event in myocardial infarction is often caused by rupture of smaller and less occlusive plaques Citation[1–3]. It has been suggested that angiogenesis plays a part in the development and destabilization of atherosclerotic plaque, but there are still matters that remain uncertain and contradictory. Some important questions that arise are the following: When does an atherosclerotic plaque become unstable? Do angiogenesis and its important mediator vascular endothelial growth factor (VEGF) enter into this, and if so what is their role? The advent of inflammatory angiogenesis in plaque formation and destabilization has recently extensively been reviewed and published in this journal Citation[4]. In their review the authors discuss the beneficial effects of neovascularization (prevention of cellular death due to better supply of oxygen and nutrients) leading to plaque growth and stabilization and on the other hand its deleterious effects, intraplaque hemorrhage and inflammation-related plaque rupture. In the present review we discuss the potential protective as well as pathophysiologic roles of VEGF as a mediator in the plaque destabilization process using the data available so far from experimental as well as clinical studies.
Atherosclerosis and unstable plaques
Although many factors are known for their influence on the development of atherosclerotic plaque, they all seem to originate from a general inflammatory process of the vascular endothelium. The underlying cause of the inflammation remains unclear, but an important and broadly supported theory is the so-called ‘response to injury theory’ Citation[5]. This hypothesis assumes that vessel wall damage with ensuing dysfunction of the endothelium is the first step in the atherosclerotic process. Factors identified as damaging to the endothelium include hypertension, diabetes mellitus, elevated low-density cholesterol levels, free radicals resulting from cigarette smoke, genetic abnormalities (e.g. elevated homocysteine plasma levels), various infections (e.g. herpes viruses and Chlamydia pneumoniae) or a combination of these factors Citation[6–9]. Endothelial dysfunction can be characterized by a change in the adhesion ability of leucocytes and thrombocytes and an increased permeability of the vascular endothelium. On top of that, the vascular endothelium's procoagulant features become more prominent and will produce a variety of cytokines and growth factors. As these processes advance, inflammatory cells will produce more and more proteins (cytokines, proteases, coagulation factors, and various vasoactive substances), of which metalloproteinases and cysteine proteinases play important roles. It can be argued that their fundamental role is to dispose of unwanted tissue components, but in turn this may result in plaque destabilization Citation[2], Citation[3], Citation[10–13]. These enzymes are capable of slowing the growth process of the fibrous cap and even dissolving collagen fibers inside the cap. As a result, the plaque destabilizes, and the fibrous cap becomes more susceptible to rupture. In addition to these basic inflammatory processes, another such process is of importance. As the plaque grows, nutrition is required and vessel growth from the adventitia takes place. It has been shown that these neovessels growing into the plaque through angiogenesis are dysmorph and immature, comparable to those in tumors and healing wounds Citation[14]. These immature vessels often leak and could contribute directly (through intraplaque hemorrhage) or indirectly (by supplying inflammatory cells) to the instability of the plaque Citation[15–17]. Angiogenesis and its mediator VEGF could accordingly take part in the atherosclerotic process.
Key messages
Angiogenesis is a normal response to ischemia, salvaging tissue at risk and preserving organ function.
Vascular endothelial growth factor (VEGF) is an important factor for maintenance of endothelial integrity and endothelial function.
Angiogenesis can also have detrimental effects if triggered by ischemia in a pathologic process such as a tumor or an atherosclerotic plaque.
Angiogenesis and VEGF seem to play a pivotal role in the development of the vulnerable atherosclerotic plaque.
VEGF
There are six VEGF variants, each with structurally similar proteins () involved in the regulation and differentiation of the vascular system, particularly in the blood and lymph vessels Citation[18–21]. Of these six subtypes VEGF-A has a major role in mediating angiogenic effects. In addition, placental growth factor (PlGF) is another member of the VEGF family. It binds to VEGFR-1. Finally, VEGF-A exists in different isoforms, which affects its heparin-binding capacity. In this review we will focus on VEGF-A and its corresponding VEGF receptors (VEGFR-1 and VEGFR-2). VEGF also plays an important role in tumor growth and metastasis; however, this topic as well as other angiogenic factors (such as the fibroblast growth factors, hepatocyte growth factor, and interleukin-8) are beyond the scope of this review.
Figure 1. Schematic representation of the different members of the vascular endothelial growth factor (VEGF) family and their corresponding receptors. PlGF = placental growth factor; VEGFR = VEGF receptor; VEGFR-1 also known as Flt-1 (=fms-like tyrosine kinase); VEGFR-2 also known as Flk-1 (=fetal liver kinase) and KDR (=kinase domain receptor); VEGFR-3 also known as Flt-4.
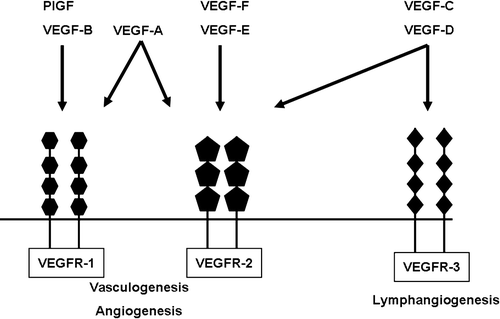
VEGF-A binds to and activates two related receptors on the cell membrane of endothelial cells, namely VEGF receptor 1 (also VEGFR-1 or Flt-1) and VEGF receptor 2 (also VEGFR-2, Flk-1, or KDR) Citation[22], Citation[23]. These receptors regulate physiological as well as pathological angiogenesis. VEGFR-2 is mainly associated with pathological angiogenesis, such as vessel formation in tumors and diabetic retinopathy. VEGFR-1, however, plays a dual role: in the embryo it has a negative influence on angiogenesis through isolation of VEGF-A; in adults it has a major effect on endothelial cells and monocytes, which stimulate angiogenesis. In addition to its indispensable role in development of the embryo, angiogenesis is essential in tissue restoration and functioning of the reproductive system Citation[24].
Functions of VEGF
VEGF is able to stimulate the proliferation and growth of endothelial cells, a process which not only occurs in arteries, but also in veins and lymph vessels. In vitro models show that VEGF induces angiogenesis in collagen gel through migration and proliferation of endothelial cells Citation[25]. Furthermore, there are several in vivo studies that show VEGF-stimulated angiogenesis in animal corneas and irises Citation[26], Citation[27]. VEGF also functions as a survival factor for endothelial cells: it can prevent apoptosis of ischemic endothelial cells. Moreover, VEGF causes the expression of various antiapoptotic proteins Citation[28], Citation[29]. VEGF is active in bone marrow as well; it influences various blood cells and appears to have hemopoietic effects including colony formation of progenitor cells of granulocytes and macrophages Citation[30]. Finally, VEGF mediates vascular permeability and several hemodynamic effects. It is capable of increasing vascular permeability and thus plays a role in inflammation and other pathological processes Citation[31]. However, is its effect on vascular permeability related to its ability to trigger angiogenesis? Although it could be an essential step in the migration and proliferation of endothelial cells Citation[32], there are examples of other proangiogenic substances which have no effect on vascular permeability but still cause strong angiogenic effects. On top of which, angiogenesis does not necessarily follow vascular leakage. In diabetic retinopathy for instance, formation of neovessels arises even decades after the first changes in vascular permeability Citation[33]. Many uncertainties still exist in this area.
Although VEGF has an important role in various physiological processes, the very same qualities cause it to play a part in the origin and maintenance of various pathological processes, including atherosclerosis.
Role of hypoxia in VEGF and atherosclerosis
The following question arises: what causes the activation of proangiogenic substances like VEGF and their receptors? Previous research on angiogenesis in tumors shows that as a tumor grows, hypoxia is a key factor for angiogenesis induction Citation[34]. The same phenomenon occurs in the growing atherosclerotic plaque. Most neovessels in the plaque originate from branches of vasa vasorum Citation[35]. As the intima of an atherosclerotic vessel thickens, a consequence of a growing plaque, oxygen diffusion from the lumen becomes more difficult. The growing of the intima to a crucial thickness (about 100 µm) can lead to a low oxygen tension and to the activation of proangiogenic substances, of which hypoxia inducible factor (HIF)-1 is one of the most important. HIF is produced in hypoxic conditions in almost all tissues, physiologically as well as pathologically Citation[36]. The HIF-1 transcription factor consists of two subunits; HIF-1β and HIF-1α, the latter of which is responsive to hypoxia Citation[37]. Under physiological (normoxic) conditions HIF-1α is modified (hydroxylated) by prolyl hydroxylases. This process is oxygen-dependent and makes use of such substances as vitamin C and iron. After HIF-1α is hydroxylated, it is further degraded by proteases. Under hypoxic conditions, however, the prolyl hydroxylases become inactive, and HIF-1α can form a dimer with HIF-1β and activate transcription of various genes, such as nitrous-oxide-synthase and VEGF Citation[34], Citation[38].
Hypoxic cells not only secrete VEGF, they also influence VEGF receptors through paracrine stimulation. As previously mentioned, two VEGF receptors have been found on endothelial cells, KDR Citation[22] and Flt-1 Citation[23], which are both tyrosine kinase receptors. In hypoxic conditions, the number of VEGF receptors will increase, facilitating angiogenic processes and enabling endothelial cells to stimulate the receptors through autophosphorylation Citation[39]. An obvious question which arises is whether or not hypoxia is the solitary cause of neovessel growth in atherosclerotic plaques. Proangiogenic substances such as VEGF also have an effect on inflammatory factors and cells. As mentioned before, unstable atherosclerotic plaques are characterized by the presence of inflammatory cells and a thin fibrous cap. What exactly is the relationship between VEGF and the inflammatory cells that abound in unstable atherosclerotic lesions?
Relationship between inflammatory processes, VEGF, and atherosclerosis
By increasing vascular permeability, VEGF could potentially play a role in the recruitment of leucocytes. Neovessels that have been formed under VEGF's influence could also contribute to leucocyte recruitment. It appears that the expressions of various adhesion molecules (for example vascular cell adhesion molecule-1) is 2–3-fold higher in endothelial cells of newly formed vessels than in normal arterial endothelium, indicating that VEGF and angiogenesis could play a large role in the leucocyte infiltration in atherosclerotic plaque Citation[40–42].
There also exists histological evidence for the infiltration of macrophages in atherosclerotic plaques by means of atherosclerotic angiogenesis Citation[43]. Previous studies show that recruitment of monocytes occurs early in the atherosclerotic process and decreases during growth of the plaque, during which proliferation of macrophages is more important Citation[44], Citation[45]. This proliferation is likely due to a previous influx of monocytes and occurs mostly in plaques with a VEGF overexpression. The attraction of circulating monocytes can be mediated by the activity of VEGF on Flt-1-positive monocytes, in addition to the increasing vascular permeability Citation[46]. From the instant that VEGF binds to the Flt-1 receptor, the expression of inflammatory factors and cytokines changes: the expression of tumor necrosis factor (TNF)-α and interleukin (IL)-1β in monocytes increases, whereas the expression of monocyte chemoattractant protein (MCP)-1 and IL-6 decreases. This could be a possible explanation for the paracrine activities (such as increased vascular permeability) of endothelial cells under influence of VEGF Citation[47–49]. Next to inflammatory factors, the activated monocytes also produce growth factors such as b-fibroblast growth factor (b-FGF), demonstrating that VEGF also causes endothelial proliferation indirectly Citation[50]. Recruited leucocytes also produce various metalloproteinases. Although these enzymes, from a physiological perspective, clear the road for growing neovessels, they also contribute as previously discussed to the weakening of the fibrous cap and thus to the instability of the plaque Citation[51], Citation[52].
Recombinant human VEGF treatment causes an increase in plaque growth. This was accompanied by increased neovascularization as well as increased macrophage density in the plaques Citation[53]. The investigators discuss that the increased vascularity in the plaques is more likely due to indirect factors such as plaque progression and macrophage recruitment than to a direct angiogenic effect Citation[53]. Interestingly, inhibition of the VEGFR-1 (Flt-1) with specific receptor-blocking antibodies reduced atherosclerotic plaque formation in ApoE-/- (apolipoprotein E knock-out) mice without reducing angiogenesis in the atherosclerotic plaques Citation[54]. In the same experiment the authors showed that inhibition of VEGFR-2 (Flk-1) did not have this antiatherogenic effect. They elegantly showed that VEGFR-1 plays an important role through the recruitment of bone marrow-derived cells not only in atherogenesis but also in arthritis and tumor formation.
It is not exclusively the Flt-1 (VEGFR-1) receptors but also the VEGFR-3 (Flt-4) receptor that play a part in neointimal inflammatory processes in atherosclerosis. A recent study on the expression of Flt-4 in atherosclerotic plaques shows that these receptors are present on macrophages and bind to VEGF-C and VEGF-D. These subtypes of VEGF (mostly involved in lymph angiogenesis) mediate apoptosis of macrophages, contributing to plaque instability Citation[55].
Increased vascular permeability also causes extravasation of erythrocytes, which in turn leads to intraplaque hemorrhage and accelerates atherosclerosis. Foam cells found in plaques regularly contain thrombocytes and erythrocytes and their waste products (glycoprotein Ib-α, free cholesterol, hemoglobin, iron) indicating that intraplaque hemorrhage leads to phagocytosis of the aforementioned cells. Because of this, iron deposition, macrophage activation, foam cell formation, and lipid accumulation arise, factors characteristic of unstable atherosclerotic plaque Citation[17], Citation[56].
VEGF is not only able to cause expression of various factors, these factors in turn can induce the expression of VEGF mRNA and in this way contribute to the abundance of VEGF in plaques. Among these factors are IL-1β, transforming growth factor (TGF)-β, and platelet-derived growth factor (PDGF)-B. Since the production of IL-1β is amplified indirectly via monocyte pathways, these mechanisms will continuously influence each other. This emphasizes the powerful role that VEGF has in angiogenesis and vascular permeability of various inflammatory processes, such as atherosclerosis Citation[57], Citation[58].
VEGF and re-endothelialization
Endothelial cells at the inner surface of the blood vessels have, in normal conditions, multiple antiatherogenic effects. These include prevention of leucocyte adhesion and thrombocyte aggregation, as well as inhibition of smooth muscle cell proliferation. One important mediator in these processes is nitric oxide (NO). VEGF is important for the maintenance of endothelial integrity. It upregulates endothelial NO synthase (eNOS) and stimulates NO synthesis. Interestingly, gene therapy with VEGF, undergone after balloon injury-accelerated re-endothelialization, reduced neointima formation and improved endothelium-dependent vasomotor function Citation[59]. Furthermore, it has been shown that VEGF-C reduced neointima formation after stenting Citation[60]. Even after adventitial transfection with VEGF gene therapy no adverse effects on restenosis were observed despite increased adventitial neovascularization Citation[61]. Finally, no adverse effects with respect to myocardial infarction or unstable angina pectoris have been reported in the numerous clinical trials with VEGF gene therapy Citation[62–66]. VEGF gene therapy has even been shown to improve coronary vasomotor function in patients with end stage coronary artery disease Citation[67].
Discussion
We have discussed that VEGF can influence many steps in the development and maintenance of the atherosclerotic plaque. On the other hand VEGF is important for maintenance of endothelial integrity and function, key both in the reduction of vascular smooth muscle cell proliferation as well as inflammatory and thrombogenic activity. The question remains as to whether or not there is experimental and clinical evidence to support VEGF as a pivotal factor in the development of plaque instability?
Various studies, both animal and human, have described proangiogenic substances and neovessels in unstable and ruptured plaques. In this context experimental studies have shown that inhibition of the VEGF-system reduces atherosclerosis. In mouse models it was demonstrated that inhibition of VEGF receptors leads to reduction of atherosclerotic lesions, independent of hypercholesterolemia Citation[68] or even angiogenesis Citation[54]. On the other hand local gene therapy with different variants of VEGF has been shown to reduce neointima formation after stent placement Citation[59] and does not have a proatherogenic effect in hypercholesterolemic mice Citation[69].
In cholesterol-fed rabbits a close relationship between angiogenesis and inflammatory processes concerning atherosclerosis was found Citation[44]. In this study, angiogenesis was inhibited with angiostatin therapy which was accompanied by a reduction in macrophage count. Interestingly, leucocyte recruitment and monocyte activation was not reduced, which emphasizes yet again the complex interaction between VEGF and inflammatory processes.
Anti-VEGF antibodies (such as bevacizumab) delay neointimal vascular formation: in plaques of cholesterol-fed rabbits treated with bevacizumab, the intimae were found to be significantly thinner than in untreated lesions Citation[70].
Stimulation of the VEGF system and the extent of plaque growth and instability has been the subject of various studies. For example, plaques were growing significantly faster in the apolipoprotein-E knock-out (ApoE-/-) mice Citation[47] than in the control group, and the number of macrophages was increased. The results in hypercholesterolemic rabbits are somewhat controversial Citation[44]. Another study on ApoE knock-out mice showed that plaques grow faster on sites where VEGF-A was administered, probably because of increased monocyte adhesion and independent of angiogenesis Citation[71].
Although the aforementioned processes are difficult to prove, these studies do show that VEGF influences growth and the extent of inflammation of atherosclerotic plaques. The question remains: what is the influence of VEGF's most basal function, angiogenesis, on atherosclerosis? Are there more neovessels found in plaques than in normal vascular endothelium?
In a human study on the distribution of vasa vasorum, a relation has emerged between the vasa vasorum density and the susceptibility of atherosclerosis in various vessel walls. For instance, a higher vasa vasorum density is found in the coronary arteries than in peripheral arteries, where atherosclerosis may be less prominent Citation[72].
Studies on the carotid arteries show links between angiogenesis and atherosclerosis as well. In a human study on symptomatic plaques in the carotids, both abnormal and immature neovessels were found, probably contributing to plaque instability Citation[17].
Nevertheless, neovessels are not found in every plaque; two other human studies on neointimal vessel formation showed that neovessels in the intima are found in restenotic lesions more frequently than in primary lesions (60% versus 40%) Citation[35], Citation[73]. The former study shows, however, that neovessel formation is more abundant in inflammatory infiltrated plaques (unstable plaques) than in stable, calcified plaques Citation[35].
There is also evidence that the amount of vasa vasorum in pathological vessels is not always different from that in normal, healthy arteries and veins. However, VEGF expression seems increased in pathological vessels Citation[74]. This study, too, shows that VEGF may have an effect on plaque growth and instability through angiogenesis-independent processes Citation[74].
To conclude, evidence for the role of angiogenesis as well as VEGF in maintaining and destabilizing atherosclerotic plaques seems to be substantial. However, precise, individual processes remain difficult to prove in vivo. Therefore it remains to be elucidated whether the influence of VEGF on plaque growth and instability is based on a direct effect on angiogenesis or indirectly through various inflammatory processes. VEGF may also prove to exert protective effects by maintaining endothelial integrity and endothelial function. More insights into the process of angiogenesis and VEGF-related mechanisms may give rise to new opportunities for diagnostic as well as therapeutic strategies. Imaging of unstable plaques by means of angiogenic tracers as well as antiangiogenic therapy have the potential to be major contributions in the combat against cardiovascular disease in the future Citation[75], Citation[76].
Acknowledgements
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006; 47(8 Suppl)C13–18
- Hackett D, Davies G, Maseri A. Pre-existing coronary stenoses in patients with first myocardial infarction are not necessarily severe. Eur Heart J. 1988; 9: 1317–23
- Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995; 92: 1084–8
- Ribatti D, Levi-Schaffer F, Kovanen PT. Inflammatory angiogenesis in atherogenesis—a double-edged sword. Ann Med. 2008; 40: 606–21
- Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999; 138: S419–20
- Ross R, Glomset JA. The pathogenesis of atherosclerosis (first of two parts). N Engl J Med. 1976; 295: 369–77
- Ross R. The pathogenesis of atherosclerosis—an update. N Engl J Med. 1986; 314: 488–500
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993; 362: 801–9
- Ross R. George Lyman Duff Memorial Lecture. Atherosclerosis: a problem of the biology of arterial wall cells and their interactions with blood components. Arteriosclerosis. 1981; 1: 293–311
- Hayden MR, Tyagi SC. Arteriogenesis: angiogenesis within unstable atherosclerotic plaque—interactions with extracellular matrix. Curr Interv Cardiol Rep. 2000; 2: 218–27
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005; 352: 1685–95
- Hartung D, Schafers M, Fujimoto S, Levkau B, Narula N, Kopka K, et al. Targeting of matrix metalloproteinase activation for noninvasive detection of vulnerable atherosclerotic lesions. Eur J Nucl Med Mol Imaging. 2007; 34(Suppl 1)S1–8
- van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994; 89: 36–44
- Khatri JJ, Johnson C, Magid R, Lessner SM, Laude KM, Dikalov SI, et al. Vascular oxidant stress enhances progression and angiogenesis of experimental atheroma. Circulation. 2004; 109: 520–5
- Khurana R, Simons M, Martin JF, Zachary IC. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005; 112: 1813–24
- Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005; 25: 2054–61
- Dunmore BJ, McCarthy MJ, Naylor AR, Brindle NP. Carotid plaque instability and ischemic symptoms are linked to immaturity of microvessels within plaques. J Vasc Surg. 2007; 45: 155–9
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004; 25: 581–611
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005; 23: 1011–27
- Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997; 57: 963–9
- Stimpfl M, Tong D, Fasching B, Schuster E, Obermair A, Leodolter S, et al. Vascular endothelial growth factor splice variants and their prognostic value in breast and ovarian cancer. Clin Cancer Res. 2002; 8: 2253–9
- Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 1992; 187: 1579–86
- de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992; 255: 989–91
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995; 1: 27–31
- Nicosia RF, Nicosia SV, Smith M. Vascular endothelial growth factor, platelet-derived growth factor, and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. Am J Pathol. 1994; 145: 1023–9
- Phillips GD, Stone AM, Jones BD, Schultz JC, Whitehead RA, Knighton DR. Vascular endothelial growth factor (rhVEGF165) stimulates direct angiogenesis in the rabbit cornea. In Vivo. 1994; 8: 961–5
- Tolentino MJ, Miller JW, Gragoudas ES, Chatzistefanou K, Ferrara N, Adamis AP. Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Arch Ophthalmol. 1996; 114: 964–70
- Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998; 273: 13313–6
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3'-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998; 273: 30336–43
- Broxmeyer HE, Cooper S, Li ZH, Lu L, Song HY, Kwon BS, et al. Myeloid progenitor cell regulatory effects of vascular endothelial cell growth factor. Int J Hematol. 1995; 62: 203–15
- Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995; 146: 1029–39
- Dvorak HF, Harvey VS, Estrella P, Brown LF, McDonagh J, Dvorak AM. Fibrin containing gels induce angiogenesis. Implications for tumor stroma generation and wound healing. Lab Invest. 1987; 57: 673–86
- Murata T, Ishibashi T, Inomata H. Immunohistochemical detection of extravasated fibrinogen (fibrin) in human diabetic retina. Graefes Arch Clin Exp Ophthalmol. 1992; 230: 428–31
- Paul SA, Simons JW, Mabjeesh NJ. HIF at the crossroads between ischemia and carcinogenesis. J Cell Physiol. 2004; 200: 20–30
- Kumamoto M, Nakashima Y, Sueishi K. Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol. 1995; 26: 450–6
- Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol. 2001; 280: C1367–74
- Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol. 2000; 59: 47–53
- North S, Moenner M, Bikfalvi A. Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors. Cancer Lett. 2005; 218: 1–14
- Namiki A, Brogi E, Kearney M, Kim EA, Wu T, Couffinhal T, et al. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem. 1995; 270: 31189–95
- de Boer OJ, van der Wal AC, Teeling P, Becker AE. Leucocyte recruitment in rupture prone regions of lipid-rich plaques: a prominent role for neovascularization?. Cardiovasc Res. 1999; 41: 443–9
- O'Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993; 92: 945–51
- O'Brien KD, McDonald TO, Chait A, Allen MD, Alpers CE. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996; 93: 672–82
- Moreno PR, Fuster V. New aspects in the pathogenesis of diabetic atherothrombosis. J Am Coll Cardiol. 2004; 44: 2293–300
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003; 100: 4736–41
- Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am J Pathol. 2002; 160: 2145–55
- Zhao Q, Egashira K, Inoue S, Usui M, Kitamoto S, Ni W, et al. Vascular endothelial growth factor is necessary in the development of arteriosclerosis by recruiting/activating monocytes in a rat model of long-term inhibition of nitric oxide synthesis. Circulation. 2002; 105: 1110–5
- Vlassara H, Fuh H, Donnelly T, Cybulsky M. Advanced glycation endproducts promote adhesion molecule (VCAM-1, ICAM-1) expression and atheroma formation in normal rabbits. Mol Med. 1995; 1: 447–56
- Wuttge DM, Sirsjo A, Eriksson P, Stemme S. Gene expression in atherosclerotic lesion of ApoE deficient mice. Mol Med. 2001; 7: 383–92
- Sengoelge G, Fodinger M, Skoupy S, Ferrara I, Zangerle C, Rogy M, et al. Endothelial cell adhesion molecule and PMNL response to inflammatory stimuli and AGE-modified fibronectin. Kidney Int. 1998; 54: 1637–51
- Pakala R, Watanabe T, Benedict CR. Induction of endothelial cell proliferation by angiogenic factors released by activated monocytes. Cardiovasc Radiat Med. 2002; 3: 95–101
- McCarthy MJ, Loftus IM, Thompson MM, Jones L, London NJ, Bell PR, et al. Angiogenesis and the atherosclerotic carotid plaque: an association between symptomatology and plaque morphology. J Vasc Surg. 1999; 30: 261–8
- Tenaglia AN, Buda AJ, Wilkins RG, Barron MK, Jeffords PR, Vo K, et al. Levels of expression of P-selectin, E-selectin, and intercellular adhesion molecule-1 in coronary atherectomy specimens from patients with stable and unstable angina pectoris. Am J Cardiol. 1997; 79: 742–7
- Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001; 7: 425–9
- Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002; 8: 831–40
- Schmeisser A, Christoph M, Augstein A, Marquetant R, Kasper M, Braun-Dullaeus RC, et al. Apoptosis of human macrophages by Flt-4 signaling: implications for atherosclerotic plaque pathology. Cardiovasc Res. 2006; 71: 774–84
- Kockx MM, Cromheeke KM, Knaapen MW, Bosmans JM, De Meyer GR, Herman AG, et al. Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2003; 23: 440–6
- Ben-Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: a potential mechanism for inflammatory angiogenesis. FEBS Lett. 1995; 372: 83–7
- Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ. Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem. 1996; 271: 736–41
- Asahara T, Chen D, Tsurumi Y, Kearney M, Rossow S, Passeri J, et al. Accelerated restitution of endothelial integrity and endothelium-dependent function after phVEGF165 gene transfer. Circulation. 1996; 94: 3291–302
- Walter DH, Cejna M, Diaz-Sandoval L, Willis S, Kirkwood L, Stratford PW, et al. Local gene transfer of phVEGF-2 plasmid by gene-eluting stents: an alternative strategy for inhibition of restenosis. Circulation. 2004; 110: 36–45
- Pels K, Deiner C, Coupland SE, Noutsias M, Sutter AP, Schultheiss HP, et al. Effect of adventitial VEGF(165) gene transfer on vascular thickening after coronary artery balloon injury. Cardiovasc Res. 2003; 60: 664–72
- Fortuin FD, Vale P, Losordo DW, Symes J, DeLaria GA, Tyner JJ, et al. One-year follow-up of direct myocardial gene transfer of vascular endothelial growth factor-2 using naked plasmid deoxyribonucleic acid by way of thoracotomy in no-option patients. Am J Cardiol. 2003; 92: 436–9
- Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, et al. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation. 2002; 105: 2012–8
- Tio RA, Tan ES, Jessurun GA, Veeger N, Jager PL, Slart RH, et al. PET for evaluation of differential myocardial perfusion dynamics after VEGF gene therapy and laser therapy in end-stage coronary artery disease. J Nucl Med. 2004; 45: 1437–43
- Laitinen M, Hartikainen J, Hiltunen MO, Eränen J, Kiviniemi M, Närvänen O, et al. Catheter-mediated vascular endothelial growth factor gene transfer to human coronary arteries after angioplasty. Hum Gene Ther. 2000; 11: 263–70
- Kastrup, J, Jørgensen, E, Rück, A, Tägil, K, Glogar, D, Ruzyllo, W,.; Euroinject One Group, et al. Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: the Euroinject One trial. J Am Coll Cardiol. 2005;45:982–8.
- Tio RA, Wijpkema JS, Tan ES, Asselbergs FW, Hospers GA, Jessurun GA, et al. Functional characteristics of coronary vasomotor function following intramyocardial gene therapy with naked DNA encoding for vascular endothelial growth factor165. Endothelium. 2005; 12: 103–6
- Petrovan RJ, Kaplan CD, Reisfeld RA, Curtiss LK. DNA vaccination against VEGF receptor 2 reduces atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007; 27: 1095–100
- Leppänen P, Koota S, Kholová I, Koponen J, Fieber C, Eriksson U, et al. Gene transfers of vascular endothelial growth factor-A, vascular endothelial growth factor-B, vascular endothelial growth factor-C, and vascular endothelial growth factor-D have no effects on atherosclerosis in hypercholesterolemic low-density lipoprotein-receptor/apolipoprotein B48-deficient mice. Circulation. 2005; 112: 1347–52
- Stefanadis C, Toutouzas K, Stefanadi E, Lazaris A, Patsouris E, Kipshidze N. Inhibition of plaque neovascularization and intimal hyperplasia by specific targeting vascular endothelial growth factor with bevacizumab-eluting stent: an experimental study. Atherosclerosis. 2007; 195: 269–76
- Lucerna M, Zernecke A, de Nooijer R, de Jager SC, Bot I, van der Lans C, et al. Vascular endothelial growth factor-A induces plaque expansion in ApoE knock-out mice by promoting de novo leukocyte recruitment. Blood. 2007; 109: 122–9
- Hildebrandt HA, Goessl M, Mannheim D, Versari D, Herrmann J, Spendlove D, et al. Differential distribution of vasa vasorum in different vascular beds in humans. Atherosclerosis. 2008; 199: 47–54
- O'Brien ER, Garvin MR, Dev R, Stewart DK, Hinohara T, Simpson JB, et al. Angiogenesis in human coronary atherosclerotic plaques. Am J Pathol. 1994; 145: 883–94
- Couffinhal T, Kearney M, Witzenbichler B, Chen D, Murohara T, Losordo DW, et al. Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) in normal and atherosclerotic human arteries. Am J Pathol. 1997; 150: 1673–85
- Jain RK, Finn AV, Kolodgie FD, Gold HK, Virmani R. Antiangiogenic therapy for normalization of atherosclerotic plaque vasculature: a potential strategy for plaque stabilization. Nat Clin Pract Cardiovasc Med. 2007; 4: 491–502
- Winter PM, Morawski AM, Caruthers SD, Fuhrhop RW, Zhang H, Williams TA, et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with alpha(v)beta3-integrin-targeted nanoparticles. Circulation. 2003; 108: 2270–4