Abstract
Background and aims. The expression of disintegrin and metalloprotease ADAM-9, ADAM-15, and ADAM-17 has been associated with cell-cell, cell-platelet, and cell-matrix interactions and inflammation. They are possibly implicated in the pathophysiology of atherosclerosis.
Methods and results. Whole-genome expression array and quantitative real-time polymerase chain reaction (PCR) analysis confirmed that ADAM-9, ADAM-15, and ADAM-17 are upregulated in advanced human atherosclerotic lesions in samples from carotid, aortic, and femoral territories compared to samples from internal thoracic artery (ITA) free of atherosclerotic plaques. Western analysis indicated that the majority of these ADAMs were in the catalytically active form. ADAM-9, ADAM-15, and ADAM-17-expressing cells were shown to co-localize with CD68-positive cells of monocytic origin in the atherosclerotic plaques using immunohistochemistry and double-staining immunofluorescence analysis. Co-localization was demonstrated in all vascular territories. In the carotid territory, cells expressing the ADAMs co-distributed also with smooth muscle cells and, in femoral territory, with CD31-positive endothelial cells, indicating that the ADAM expression pattern depends on vascular bed territory.
Conclusions. Present findings provide strong evidence for the involvement of catalytically active ADAM-9, ADAM-15, and ADAM-17 in advanced atherosclerosis, most notably associated with cells of monocytic origin.
Introduction
The inflammatory and chronic nature of atherosclerosis Citation[1] makes it compelling to identify genes involved in this process. To better understand the alterations in gene expression in advanced atherosclerosis, we utilized genome-wide expression array (GWEA), encompassing all the known 23,000 genes to study a unique sample material of advanced plaques in aorta, carotid and femoral arteries, and control samples from internal thoracic artery (ITA) classified according to the American Heart Association classification Citation[2]. Over 200 genes were found to be upregulated, including members of the disintegrin and metalloprotease (ADAM) family Citation[3]. Of these, ADAM-9, ADAM-15, and ADAM-17 were selected for more detailed analysis because they have been associated with cellular and physiological functions pertinent to atherosclerosis, such as cell-cell, cell-platelet, and cell-matrix interactions, as well as with inflammation Citation[4], Citation[5].
Key messages
Upregulated expression of ADAM-9, ADAM-15, and ADAM-17 and the presence of their catalytically active forms are implicated in advanced human atherosclerosis and suggest roles in the monocyte homing, migration, or proliferation in aorta, carotid, and femoral arteries.
As the therapeutic intervention of ADAMs in human diseases is actively pursued, our findings underline the potential of targeting the development of atherosclerosis through modulation of ADAM function.
A precise picture of ADAMs expression is a prerequisite for understanding the contribution of their intricate interplay in atherosclerosis, towards which the present study takes an important step.
ADAM metalloprotease disintegrins are transmembrane proteins mediating targeted proteolysis, cell adhesion, and signal transduction Citation[3], Citation[6]. ADAM proteinases can release and activate cytokines, growth factors, and other bioactive proteins from their membrane-bound precursors and remove receptors from the cell surface, in the process called ectodomain shedding Citation[4], Citation[5]. However, not all ADAMs are active proteinases as half of them do not contain all the essential amino acids at the active site Citation[5]. ADAMs have also been implicated in cell-cell and cell-matrix interactions through binding to integrins and other adhesion molecules Citation[6]. Their diverse involvement in cell interactions suggests that alterations in the expression of ADAMs may play roles in complex pathologies such as atherosclerosis.
ADAM-9 has been implicated both in ectodomain shedding and cell interactions through integrin binding. The potential ADAM-9 sheddase substrates include growth factors and cytokines Citation[5] which have been linked to atherosclerosis Citation[7], Citation[8]. ADAM-9 is capable of binding to α6β1 and αvβ5Citation[9], Citation[10], and also these interactions might be relevant in atherosclerosis. ADAM-9 interaction with α6β1 was shown to regulate fibroblast motility, and α6β1 can induce the mobility of several cell types including macrophages Citation[11–13]. The interaction with αvβ5 was shown to stimulate proinflammatory interleukin-6 production in cultured osteoblasts Citation[10]. ADAM-9 has indeed been shown to be expressed in human atherosclerotic plaques, in conjunction with increased levels of αvβ3 and α5β1 integrins which also are associated with the development and progression of atherosclerosis Citation[14]. No ADAM-9 nor αvβ3 or α5β1 mRNA and only weak ADAM-9 immunostaining were detected from human thyroid artery without atherosclerosis Citation[14]. Altogether, these interactions and expression data are consistent with a potential role of ADAM-9 in atherosclerosis.
The first direct indication of a potential role of an ADAM in atherosclerosis was, indeed, the demonstration of upregulated ADAM-15 in monkey atherosclerotic lesions Citation[1]. Also ADAM-15 has been shown to mediate both ectodomain shedding and binding to integrins. The potential sheddase substrates of ADAM-15 include epidermal growth factor (EGF) family growth factors Citation[5] with possible association with atherosclerosis Citation[7]. Human ADAM-15, but remarkably not rodent adam-15, contains the ‘classical’ arginine-glycine-aspartate (RGD) sequence in its putative integrin-binding motif which appears to mediate binding to α5β1, αvβ3, and α(IIb)β3 integrins Citation[9], Citation[15], Citation[16]. ADAM-15 is highly expressed in mouse vascular cells and endocardium Citation[17], and in cultured human aortic smooth muscle and umbilical vein endothelial cells Citation[1]. Similarly to ADAM-9, weak ADAM-15 immunostaining was detected from normal human arteries while the mRNA level was below detection, but atherosclerotic plaques from carotid arteries showed upregulated ADAM-15 (and ADAM-9) expression in conjunction with elevated α5β1 and αvβ3 integrins Citation[14]. ADAM-15 seems to be an important mediator of inflammation Citation[18]. Hence, also ADAM-15 is an intriguing candidate to be involved in the pathology of atherosclerosis.
ADAM-17, besides ADAM-10, is the most studied sheddase ADAM, originally identified as the tumor necrosis factor alpha (TNF-α) converting-enzyme (TACE) Citation[19], Citation[20]. In addition to TNF-α, ADAM-17 can activate several cytokines and growth factors implicated in atherosclerosis and remove their receptors from cell surfaces Citation[5], and the number of putative novel substrates continues to grow. Since ADAM-17 has been implicated in atherosclerosis Citation[21], Citation[22], and since it can modulate leukocyte adhesion by shedding L-selectin Citation[23], Citation[24] and modulating macrophage activity Citation[25], ADAM-17 is an interesting potential target for the treatment of inflammatory diseases such as atherosclerosis Citation[8]. Further investigations of ADAM-17 will also advance our understanding of the molecular basis of atherosclerosis.
The current view of possible roles of ADAMs in atherosclerosis is based mainly on animal and in vitro experiments, and on a single report of arterial location but without classification of the degree of atherosclerosis or verification of gene expression data Citation[14], Citation[22]. Based on our genome-wide expression array data on ADAMs, we hypothesized that their expression is upregulated in advanced atherosclerosis and might co-distribute with endothelial, smooth muscle, or monocytic cells in atherosclerotic areas. Thus we performed detailed analysis of atherosclerotic plaques in different arterial beds, i.e. carotid region, aorta, and femoral region, to find out whether ADAMs associate with global progression of atherosclerosis in human subjects.
Methods
Vascular samples
Vascular sample series from femoral arteries, carotid arteries, and abdominal aortas were obtained during open vascular procedures from 30 patients under surveillance of a senior consultant vascular surgeon (NO). Six control samples were taken from internal thoracic arteries (ITA) obtained during coronary artery bypass grafting. The study has been approved by the Ethics Committee of Tampere University Hospital. All clinical investigations were conducted according to declaration of Helsinki principles. The samples were taken from patients subjected to open vascular surgical procedures in the Division of Vascular Surgery and Heart Centre, Tampere University Hospital. The vascular samples were classified according to recommendation of American Heart Association (AHA) Citation[2].
RNA isolation and genome-wide expression analysis (GWEA)
The fresh tissue samples (n=26) were immediately soaked in RNALater solution (Ambion Inc., Austin, TX, USA), and total-RNA was isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA) and RNAEasy Kit (Qiagen, Valencia, CA, USA). The concentration and quality of RNA were evaluated spectrophotometrically (BioPhotometer, Eppendorf, Wesseling-Berzdorf, Germany). The GWEA microarray experiments were performed by using Sentrix® Human-8 Expression BeadChips analyzing over 23,000 known genes and gene candidates (Illumina, San Diego, CA, USA) according to given instructions by the manufacturer. In brief, a 200 ng aliquot of total-RNA from each sample was amplified to cDNA using Ambion's Illumina RNA Amplification kit following the instructions (cat. no I1755, Ambion, Inc., Austin, TX, USA). In vitro transcription (IVT) reaction of cDNA to cRNA was performed overnight (14 h) including biotin-11-deoxy uridine triphosphate (dUTP) (PerkinElmer Life And Analytical Sciences, Inc., Boston, MA, USA) for labeling the cRNA product. Both before and after the amplifications the RNA/cRNA concentrations were checked with Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA), and RNA/cRNA quality was controlled by BioRad's Experion Automated Electrophoresis System and RNA StdSens Analysis Kit (BioRad Laboratories, Inc., Hercules, CA, USA). Each sample cRNA (1500 ng) was hybridized to Illumina's Sentrix® Human-8 Expression BeadChip arrays (Illumina) at 55°C overnight (18 h) following the Illumina Whole-Genome Gene Expression Protocol for BeadStation. Hybridized biotinylated cRNA was detected with 1 µg/mL cyanine3-streptavidine (Amersham Biosciences, Pistacataway, NJ, USA). BeadChips were scanned with Illumina BeadArray Reader. The accuracy of Illumina Sentrix® Human-8 Expression BeadChips microarray methodology to measure the gene expression was earlier verified by real-time quantitative TaqMan PCR by quantitating the expression of 20 genes with both methods Citation[26].
Quantitative real-time PCR (qPCR)
From the 30 tissue samples, gene expression analyses were done with TaqMan low-density arrays (LDAs) (Applied Biosystems, Foster City, CA, USA). Total-RNA (500 ng) was reverse-transcribed to cDNA using High Capacity cDNA Kit according to the manufacturer's instructions (Applied Biosystems). For the PCR, LDAs were loaded with 8 µL undiluted cDNA, 42 µL H2O, and 50 µL PCR Universal Master Mix and run according to the manufacturer's instructions (Applied Biosystems). Samples were analyzed as duplicates, and both cDNA synthesis and PCR reactions were validated for inhibition. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene control, and the qPCR results were analyzed using SDS 2.2 Software (Applied Biosystems).
Western analysis (WB)
Four randomly selected samples of atherosclerotic plaques from carotid (two patients) and femoral artery, and aorta were homogenized (100 mg/350 µL) in ice-cold lysis buffer (10 mM Tris-HCl pH 7.4, 1% (w/v) Triton X-100, 0.1% (w/v) sodium dodecyl suphate (SDS), 0.1% (w/v) sodium deoxycholate, 150 mM NaCl) Citation[27] containing Roche Complete protease inhibitors (Roche Diagnostics, Mannheim, Germany) using a Potter-Elvehjem homogenizer with a teflon pestle. The amount of protein was not measured but the samples (each 100 mg wet weight) were prepared in equal volumes of homogenization and sample buffers. Insoluble debris was removed with centrifugation (16.2 k×g, 10 min) in a refrigerated microcentrifuge at 4°C. Laemmli SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer (5x) with dithiothreitol (DTT) was added to 1x concentration (0.1 M DTT final). The samples were heated at 99°C for 5 minutes, cooled to room temperature and centrifuged 16.2 k×g for 10 minutes prior to loading 100 µL/well onto a 10% SDS-polyacrylamide gel. Molecular weight markers (Chemichrome Ultimate, C2117, Sigma, St. Louis, MI, USA) were run along the samples. Following the SDS-PAGE, the proteins were electrotransferred onto a polyvinylidene fluoride (PVDF) membrane (BioRad, Hercules, CA, USA) in a semi-dry blotter. The membrane was then treated with 0.1% (w/v) Ponceau S in 5% acetic acid to verify the successful transfer, to ensure an even loading, and to fix the proteins onto the membrane.
The primary antibodies against human ADAM protein ectodomains were from R&D Systems (Abingdon, UK). Goat-anti-ADAM-9 (AF939), goat-anti-ADAM-15 (AF935), and goat-anti-ADAM-17 (AF2129) were diluted 1/1000 in tris-buffered saline with Tween-20 (TBST) containing 5% non-fat milk (TBS with 0.1% (w/v) Tween-20). The peroxidase-coupled secondary antibody, anti-goat/sheep (A9452, Sigma), was diluted 1/5000 in TBST/milk.
For the immunodetection, the blots were blocked with 5% (w/v) non-fat milk in TBST, followed with 1 h incubation at room temperature (RT) with the primary antibodies. After several TBST washes and the secondary antibody incubation (1 h, RT), the blots were washed with TBST, rinsed in TBS, and the bound secondary antibody was visualized with enhanced chemiluminescence (ECL) and a digital imager. Just prior to the ECL reagent incubation, the blots were rinsed in water. The ECL was done with SuperSignal West Dura reagents according to the manufacturer's protocol (Pierce, Rockford, USA). The ECL was visualized in a ChemiDoc XRS digital imager using Quantity One 4.5.2 software (BioRad).
Immunohistochemistry (IH)
Immunohistochemistry was performed using the avidin biotin complex (ABC) method (Vectastain Elite kit, Vector Laboratories, Burlingame, CA, USA) and paraffin-embedded vascular samples without any counterstain. ADAMs in vascular wall were detected with rabbit anti-human ADAM-9 (AF1031), mouse anti-human ADAM-15 (MAB935), and chicken anti-human ADAM-17 (AF930) ectodomain antibodies (R&D Systems, Minneapolis, MN, USA). The following primary antibodies were used to detect vascular cell markers in adjacent sections: Muscle actin (mouse anti-human muscle actin, clone HHF35; DakoCytomation, Glostrup, Denmark) was used to detect smooth muscle cells. CD68 (mouse anti-human CD68, clone PG-M1; DakoCytomation) was used as marker of monocytes and macrophages. For the detection of endothelial cells, CD31 antibody (mouse anti-human CD31, endothelial cell, clone JC70A; DakoCytomation) was used. The sections were subjected to microwave antigen retrieval treatment as described earlier Citation[28]. Endogenous peroxidase activity was extinguished by treating the section with 0.3% H2O2 for 30 min. Subsequently the sections were incubated overnight with the primary antibodies followed by biotinylated horse anti-goat, goat anti-chicken (1:500, Vector Laboratories), or sheep anti-mouse (1:300, Amersham Int., Buckinghamshire, UK) and ABC complex for 30 min. Diaminobenzidine was used as the chromogen. All antibodies were diluted in phosphate buffered saline (PBS) containing 1% bovine serum albumin (BSA) and 0.3% of Triton X-100. Controls included omitting the primary antibody or replacing it with non-immune sera. No staining was seen in the controls.
The co-localization of CD68 and ADAM-9 and ADAM-17 in carotid arteries was studied with double-staining immunofluorescence (IF). The samples were fixed with 4% paraformaldehyde (in 0.1 M PBS, pH 7.3) for 6 h at +4°C and cryoprotected with 20% sucrose in PBS. Frozen sections (6 µm) were cut with Micron HM560 Cryostat and thaw-mounted onto Polysine glass slides (Menzel, Braunschweig, Germany). The sections were incubated overnight with mouse monoclonal anti-CD68 (1:10) and rabbit anti-ADAM-9 (1:100), or with the mixture of CD68 antibody and chicken anti-ADAM-17 followed by a mixture of biotinylated sheep anti-mouse antibody (1:200, Amersham) and rhodamine-conjugated goat anti-rabbit antibody (1:50, Boehringer Mannheim), or with a mixture of fluorescein-labeled sheep anti-mouse antibody (1:10, Amersham) and biotinylated goat anti-chicken (Vector Labs, Peterborough, England) for 30 min at +37°C, respectively. Subsequently, the sections were incubated with fluorescein isothiocyanate (FITC)-conjugated avidin or rhodamine-conjugated avidin (1:100, Vector Labs) for 30 min, respectively. Sections were mounted in a mixture of glycerol and PBS (3:1) containing 0.1% paraphenylenediamine and examined in Nikon Microphot FXA microscope equipped with proper fluorescence filters. Photographs were obtained with a PCO Sensicam digital camera (PCO, Kelheim, Germany). Alternatively the co-localization of ADAM-9, ADAM-15, ADAM-17, and CD68 was studied in 5 µm paraffin sections (mirror image sections). Sections were stained with ABC method as described above.
Statistical analyses
Statistical analyses were performed using SPSS version 14.0 (SPSS Inc., Chicago, IL, USA). The non-parametric Mann-Whitney U-test was used for comparison of mRNA expression between atherosclerotic and control tissues and between different arterial regions. Data are presented as median and range.
Results
Characteristics of the subjects and studied vascular samples
The median age was 70.0 years (45–93 years) and 75.9% of the subjects were men. The prevalence of risk factors was as follows: dyslipidemia 40.7%, hypertension 77.8%, diabetes 18.5%, history of smoking 86.2%, alcohol usage more than once a week 42.8%. A total of 73.3% of the samples were classified as type 5–6 advanced plaques.
ADAM mRNA expression in the atherosclerotic tissue
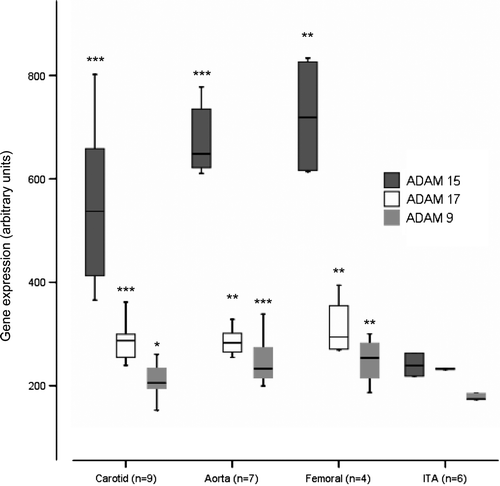
We compared the ADAM-9, ADAM-15, and ADAM-17 expression level between atherosclerotic tissue (types 5–6) and control tissue from internal thoracic artery (ITA) (type 0), and expression levels between different vessel types. In GWEA, the median ADAM-9 expression was elevated relative to ITA in the carotid arteries (1.2-fold, P = 0.012), aortas (1.3-fold, P = 0.001), and femoral arteries (1.5-fold, P = 0.010), respectively (). ADAM-15 expression was elevated relative to ITA in the carotid arteries (2.2-fold, P < 0.001), aortas (2.7-fold, P < 0.001), and femoral arteries (3.0-fold, P = 0.010), respectively (). ADAM-17 expression was elevated relative to ITA in the carotid arteries (1.2-fold, P < 0.001), aortas (1.2-fold, P = 0.001), and femoral arteries (1.3-fold, P = 0.01), respectively ().
Figure 1. ADAM-9, ADAM-15, and ADAM-17 expression in control samples from internal thoracic artery (ITA), and carotid, aortic, and femoral artery samples. Gene expression value is the normalized average gene intensity for each group measured in the Illumina Expression BeadChips. *P < 0.05, **P < 0.01, ***P < 0.001 relative to ITA, Mann-Whitney U-test.
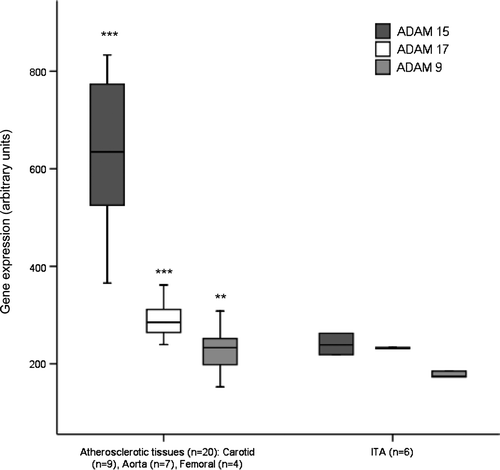
According to GWEA, the median ADAM-9 expression level in atherosclerotic tissues (n=20) was 233 (153–338) and 174 (173–185) in ITA tissues (type 0, n=6) (1.3-fold relative to ITA, P = 0.001) (). The median ADAM-15 expression level in atherosclerotic tissues (n=20) was 635 (366–834), and 239 (219–263) in ITA (type 0, n=6) (2.7-fold relative to ITA, P < 0.001) (). The median ADAM-17 expression level in atherosclerotic tissues (n=20) was 285 (239–395), and 232 (231–234) in ITA (type 0, n=6) (1.2-fold relative to ITA, P < 0.001) ().
Figure 2. ADAM-9, ADAM-15, and ADAM-17 expression in atherosclerotic samples and control samples from internal thoracic artery (ITA). Gene expression value is the normalized average gene intensity for each group measured in the Illumina Expression BeadChips. *P < 0.05, **P < 0.01, ***P < 0.001 relative to ITA, Mann-Whitney U-test.
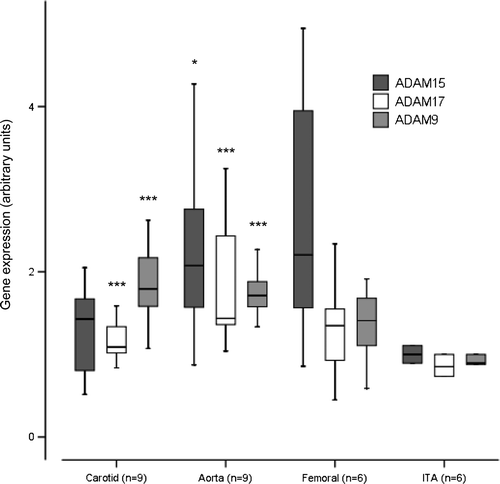
The Illumina GWEA array results were verified with quantitative PCR. ADAM-9 expression was elevated in the carotid arteries (1.8-fold, P < 0.001), aortas (1.7-fold, P < 0.001), and there was trend of increase in femoral arteries (1.4-fold, P = 0.065), respectively (). ADAM-15 expression was not elevated in the carotid arteries (1.4-fold, P = 0.607), but there was a significant increase in aortas (2.1-fold, P = 0.012), and a trend of increase in femoral arteries (2.2-fold, P = 0.065), respectively (). ADAM-17 expression was elevated in the carotid (1.1-fold, P < 0.001), aorta (1.4-fold, P < 0.001), and it was not elevated in femoral artery (1.3-fold, P = 0.132), respectively (). No apparent difference in gene expression profiles was found between early (type 5) and advanced plaques (type 6).
Figure 3. Expression of ADAM-9, ADAM-15, and ADAM-17 mRNA in control samples from internal thoracic artery (ITA), and carotid, aortic, and femoral artery samples measured with TaqMan Low-Density Array. *P < 0.05, **P < 0.01, ***P < 0.001 relative to ITA, Mann-Whitney U-test.
ADAM protein expression in the atherosclerotic tissue
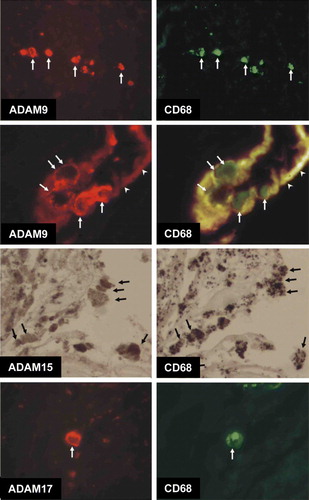
Immunohistochemistry of atherosclerotic lesions revealed that ADAM-9, ADAM-15, and ADAM-17 proteins were expressed in the atheromatous core and to some extent in all the vessel layers in early and advanced plaques in all the vessel beds while only sparse cells were positive in ITA vessels (). In aortas ADAM-9, ADAM-15, and ADAM-17-staining cells co-distributed mainly with CD68-positive cells in the plaque but did not co-distribute with CD31-positive cells (A). In femoral arteries, however, ADAM-9, ADAM-15, and ADAM-17-positive cells co-distributed with endothelial cells and in addition with macrophages (B). In the carotid arteries, ADAM-9, ADAM-15, and ADAM-17-positive cells co-distributed with both macrophages and smooth muscle cells (C). Immunofluorescence double-staining microscopy of a representative carotid plaque confirmed that ADAM-9 and ADAM-17-positive cells co-localized with CD68-positive cells (). The staining of ADAM-9 and ADAM-17 was localized at the plasma membrane while CD68 was cytoplasmic (). ADAM-15 staining co-localized with CD68 in the mirror sections (). The representative figure shows expression of ADAM-9 in macrophages invading the endothelial cell layer in a neovascularization capillary within the atherosclerotic plaque ().
Figure 4. Expression of ADAM-9, ADAM-15, and ADAM-17 in human atherosclerotic plaques. Serial staining of monocyte marker CD68 and smooth muscle cell marker HHF35 in human aortic (A), femoral (B), and carotid (C) plaques. Samples from internal thoracic artery (ITA) served as controls (D). NV indicates a neovascularized vessel. A straight line indicates the transition between the intima and media. Arrows indicate typical positively stained cells. The stage of atherosclerosis was classified according to American Heart Association (AHA) classification (type 1–6). 100× magnification.
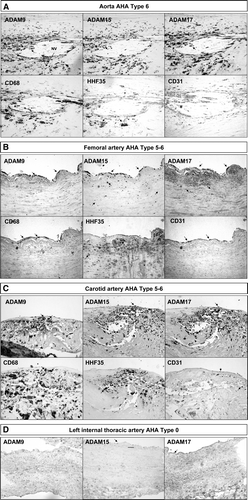
Figure 5. Double immunofluorescence images demonstrating co-localization of ADAM-9 and ADAM-17 and CD68, and mirror image sections showing the co-localization of ADAM-15 and CD68. ADAM-9-immunoreactive cells at the border of adventitia and media (arrows) are also labeled with CD68. High-magnification image showing ADAM-9/CD68-immunoreactive cells (arrows) adhered to the intima of neovascularization capillary (arrowheads point to the intima exhibiting red/yellow autofluorescence). ADAM-9 labeling is very strong in the cell membrane. Mirror image sections indicating co-localization of ADAM-15 and CD68. Arrows point to the cells immunoreactive to both antigens. Double immunofluorescence photographs demonstrating the co-localization of ADAM-17 and CD68. ADAM-17 immunoreactivity is very strong in the cell membrane whereas CD68 labeling is seen throughout the cytoplasm.
The majority of ADAM-9, ADAM-15, and ADAM-17 protein was in the catalytically active form in the atherosclerotic plaques of carotid and femoral arteries and aorta (). A putative negligible signal of unprocessed pro-form at c. 110 kDa was seen only after very long over-exposure (1 h) for ADAM-9 and ADAM-15 (data not shown). Hence, virtually all ADAM-9, ADAM-15, and ADAM-17 protein appears to be in the processed mature, presumably catalytically competent form.
Figure 6. Western detection of (A) ADAM-9, (B) ADAM-15, and (C) ADAM-17 in atherosclerotic patient samples from carotid Citation[1], Citation[3], femoral Citation[2], and aortic Citation[4] plaques, visualized with enhanced chemiluminescence. The positions of the molecular weight markers and active/inactive forms are indicated.
![Figure 6. Western detection of (A) ADAM-9, (B) ADAM-15, and (C) ADAM-17 in atherosclerotic patient samples from carotid Citation[1], Citation[3], femoral Citation[2], and aortic Citation[4] plaques, visualized with enhanced chemiluminescence. The positions of the molecular weight markers and active/inactive forms are indicated.](/cms/asset/1cdc8f33-5b11-4774-ba61-df8f88847c1e/iann_a_365143_f0006_b.gif)
Discussion
The present study provides novel evidence suggesting that the metalloprotease disintegrins ADAM-9, ADAM-15, and ADAM-17 Citation[4], Citation[5] are associated with the pathophysiology of human atherosclerosis. The results corroborate those of the earlier animal model and in vitro studies Citation[1] indicating roles for these ADAMs in atherosclerosis, in addition to the first demonstration of upregulation of ADAM-9 and ADAM-15 in human atherosclerotic carotid arteries Citation[14]. The combined examination with GWEA, qPCR, immunohistochemistry (IHC), and IF showed that ADAM-9, ADAM-15, and ADAM-17 expression was upregulated in advanced atherosclerosis at the transcriptional and protein levels. The results are novel as four different vascular territories were examined, and they suggest that the upregulated expression of these ADAMs is likely to be associated with monocytic cells in the atherosclerotic plaques and with smooth muscle and endothelial cells in the carotid and femoral territories, respectively. A precise picture of ADAMs expression is a prerequisite for understanding the contribution of their intricate interplay in atherosclerosis, towards which the present study takes an important step.
In the previous study by Al-Fakhri et al., the mRNA levels of ADAM-9 and ADAM-15 in human carotid artery plaques were increased 9- and 7-fold, respectively, as compared to thyroid artery samples from subjects free of atherosclerotic disease Citation[14]. In the present study, the fold-change was lower, which might be explained by the fact that our control samples from internal thoracic artery were from subjects with coronary artery atherosclerosis and, albeit there was no histological evidence of atherosclerosis, it is obvious that the systemic nature of atherosclerosis might be reflected in the levels of ADAMs in all the vascular beds narrowing the difference in expression levels. According to the previous study, ADAM-9 and ADAM-15 were expressed in smooth muscle cells in the neointima of atherosclerotic arteries Citation[14]. Co-localization of ADAM-9, ADAM-15, and ADAM-17-positive cells with those labeled with CD68 antibody indicates that these cells are of monocytic origin, although these ADAMs were localized also to smooth muscle cells in the carotid samples. Whether the rheologic properties of different vascular beds have a different effect on expression of ADAMs in specific cell types in the plaque remains to be elucidated.
Proteolytic processing, the detachment of the pro-domain, has been associated with the maturation of at least some ADAMs, including ADAM-9 and ADAM-17, to a catalytically active form Citation[5], Citation[6]. Western analysis indicated that virtually all ADAM-9, ADAM-15, and ADAM-17 protein was in the catalytically active form in atherosclerotic lesions in carotid and femoral arteries and in aorta and hence in the catalytically competent state.
The present findings support a role of ADAM-9 in the pathology of atherosclerosis through ectodomain shedding, cell-cell, or cell-matrix interactions. ADAM-9 has been shown to be able to cleave several substrates, including heparin binding-EGF (HB-EGF) Citation[29], and thus its upregulation might result in misregulated shedding of factors effecting atherogenesis Citation[30]. The ability of ADAM-9 to mediate cell interactions through α6β1 and αvβ5 integrins Citation[10], Citation[31] may also contribute to atherosclerosis since these interactions can regulate cell motility and the production of interleukin-6 Citation[9], Citation[10].
The co-localization of ADAM-15-expressing cells with CD68 and co-distribution with CD31-positive cells in atherosclerotic areas strongly suggest that ADAM-15 on monocytic or endothelial cells might play a role in monocyte-macrophage migration, endothelial-platelet adhesion, or matrix remodeling in atherosclerosis. ADAM-15 on endothelial cells might interact with αvβ3 integrin on leukocytes, especially monocytes, during atherogenesis. Consistently cultured hematopoietic cells of monocytic lineage express αvβ3 integrin which was shown to bind recombinant ADAM-15 Citation[31]. Also, ADAM-15 binding to αvβ3 or α5β1 on smooth muscle cells in atherogenic areas could modulate their migration/proliferation properties Citation[32]; however, we did not observe co-distribution of ADAM-15 with the smooth muscle cells. Furthermore, ADAM-15-mediated interactions between the endothelial cells and platelets are potentially important since the initiation, inflammation, atherogenesis, and plaque formation, including the recruitment of progenitor and dendritic cells, require adherence of platelets to the endothelial cells. This would be consistent with the platelet adhesion to ADAM-15-expressing endothelial-like cells and subsequent platelet activation and microthrombus formation in a flow chamber model Citation[16]. Given the inflammatory nature of atherosclerosis, it is interesting that in inflammatory bowel diseases, ADAM-15 is upregulated in epi- and endothelial cells in close contact with α5β1-expressing leukocytes, suggesting a role in leukocyte migration Citation[33]. This also corroborates our hypothesis of ADAM-15 possibly contributing to monocyte-macrophage migration in atherosclerosis.
To our knowledge, the present study provides the first comprehensive immunohistochemical examination of ADAM-17 expression in human atherosclerosis. The results agree with and extend those of the earlier investigations implicating ADAM-17 in the pathology of atherosclerosis. Previously, Canault and co-authors have demonstrated elevated ADAM-17 protein levels in the lipidic core of human atherosclerotic plaques and showed with a detailed analysis of an apolipoprotein E-deficient mouse model that ADAM-17 immunostaining increased along with the development of atherosclerotic lesions Citation[21]. In a subsequent study they provided convincing evidence indicating the pathophysiological role of ADAM-17 sheddase activity in human atherosclerosis Citation[22]. The contribution of ADAM-17 in atherosclerosis is also corroborated by the studies showing increased TNF-α levels after acute myocardial infarction Citation[34], Citation[35] and localization of TNF-α expression in atheromas Citation[36]. The ability of ADAM-17 to downregulate macrophage colony-stimulating factor receptor (M-CSFR) from macrophages undergoing activation enables fully activated phenotype in mononuclear macrophages by rapidly preventing M-CSF-mediated signaling Citation[25], suggesting that also ADAM-17 might effect macrophage function in atherogenesis. To sum up, our results combined with other studies suggest that ADAM-17 might contribute to atherosclerosis by activating and inactivating pro- and anti-inflammatory factors, respectively, and thus regulating inflammation, leukocyte adhesion, migration, and proliferation.
In conclusion, our results implicate ADAM-9, ADAM-15, and ADAM-17 in advanced atherosclerosis and suggest roles in the monocyte homing, migration, or proliferation in all major vascular beds. As the therapeutic intervention of ADAMs in human diseases is actively pursued Citation[5], Citation[37], our findings underline the potential of targeting the development of atherosclerosis through modulation of ADAM function.
Acknowledgements
The authors wish to thank Mrs Ulla Jukarainen, Nina Peltonen, and Ms Elli Ada Olivia Oksala for their skillful technical assistance. This research is funded by European Union 7th Framework Program grant number 201668, Atheroremo. This study was supported with grants from the Medical Research Fund of Tampere University Hospital, the Emil Aaltonen Foundation (T.L.), the Pirkanmaa Regional Fund of the Finnish Cultural Foundation, the Research Foundation of Orion Corporation, the Jenny and Antti Wihuri Foundation, and the Academy of Finland (Grant no. 104821). Niku Oksala was supported by grants from the Finnish Angiology association, Maire Taponen foundation, and Paavo Nurmi foundation. Niku Oksala and Mari Levula contributed equally to the work. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Herren B, Raines EW, Ross R. Expression of a disintegrin-like protein in cultured human vascular cells and in vivo. FASEB J. 1997; 11: 173–80
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995; 92: 1355–74
- Wolfsberg TG, Straight PD, Gerena RL, Huovila AP, Primakoff P, Myles DG, et al. ADAM, a widely distributed and developmentally regulated gene family encoding membrane proteins with a disintegrin and metalloprotease domain. Dev Biol. 1995; 169: 378–83
- Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005; 6: 32–43
- Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM. Shedding light on ADAM metalloproteases. Trends Biochem Sci. 2005; 30: 413–22
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003; 17: 7–30
- Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis. 2006; 186: 38–53
- Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. 2007; 48: 751–62
- Nath D, Slocombe PM, Webster A, Stephens PE, Docherty AJ, Murphy G. Meltrin gamma (ADAM-9) mediates cellular adhesion through alpha(6)beta(1)integrin, leading to a marked induction of fibroblast cell motility. J Cell Sci. 2000; 113(Pt 12)2319–28
- Karadag A, Zhou M, Croucher PI. ADAM-9 (MDC-9/meltrin-gamma), a member of the a disintegrin and metalloprotease family, regulates myeloma-cell-induced interleukin-6 production in osteoblasts by direct interaction with the alpha(v)beta5 integrin. Blood. 2006; 107: 3271–8
- Jasiulionis MG, Chammas R, Ventura AM, Travassos LR, Brentani RR. alpha6beta1-Integrin, a major cell surface carrier of beta1-6-branched oligosaccharides, mediates migration of EJ-ras-transformed fibroblasts on laminin-1 independently of its glycosylation state. Cancer Res. 1996; 56: 1682–9
- Gimond C, Baudoin C, van der Neut R, Kramer D, Calafat J, Sonnenberg A. Cre-loxP-mediated inactivation of the alpha6A integrin splice variant in vivo: evidence for a specific functional role of alpha6A in lymphocyte migration but not in heart development. J Cell Biol. 1998; 143: 253–66
- Shaw LM, Mercurio AM. Regulation of cellular interactions with laminin by integrin cytoplasmic domains: the A and B structural variants of the alpha 6 beta 1 integrin differentially modulate the adhesive strength, morphology, and migration of macrophages. Mol Biol Cell. 1994; 5: 679–90
- Al-Fakhri N, Wilhelm J, Hahn M, Heidt M, Hehrlein FW, Endisch AM, et al. Increased expression of disintegrin-metalloproteases ADAM-15 and ADAM-9 following upregulation of integrins alpha5beta1 and alphavbeta3 in atherosclerosis. J Cell Biochem. 2003; 89: 808–23
- Zhang XP, Kamata T, Yokoyama K, Puzon-McLaughlin W, Takada Y. Specific interaction of the recombinant disintegrin-like domain of MDC-15 (metargidin, ADAM-15) with integrin alphavbeta3. J Biol Chem. 1998; 273: 7345–50
- Langer H, May AE, Bultmann A, Gawaz M. ADAM 15 is an adhesion receptor for platelet GPIIb-IIIa and induces platelet activation. Thromb Haemost. 2005; 94: 555–61
- Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, et al. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol. 2003; 23: 5614–24
- Charrier-Hisamuddin L, Laboisse CL, Merlin D. ADAM-15: a metalloprotease that mediates inflammation. FASEB J. 2008; 22: 641–53
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloprotease disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997; 385: 729–33
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloprotease that processes precursor tumour-necrosis factor-alpha. Nature. 1997; 385: 733–6
- Canault M, Peiretti F, Kopp F, Bonardo B, Bonzi MF, Coudeyre JC, et al. The TNF alpha converting enzyme (TACE/ADAM17) is expressed in the atherosclerotic lesions of apolipoprotein E-deficient mice: possible contribution to elevated plasma levels of soluble TNF alpha receptors. Atherosclerosis. 2006; 187: 82–91
- Canault M, Leroyer AS, Peiretti F, Leseche G, Tedgui A, Bonardo B, et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol. 2007; 171: 1713–23
- Condon TP, Flournoy S, Sawyer GJ, Baker BF, Kishimoto TK, Bennett CF. ADAM17 but not ADAM10 mediates tumor necrosis factor-alpha and L-selectin shedding from leukocyte membranes. Antisense Nucleic Acid Drug Dev. 2001; 11: 107–16
- Gomez-Gaviro M, Dominguez-Luis M, Canchado J, Calafat J, Janssen H, Lara-Pezzi E, et al. Expression and regulation of the metalloprotease ADAM-8 during human neutrophil pathophysiological activation and its catalytic activity on L-selectin shedding. J Immunol. 2007; 178: 8053–63
- Rovida E, Paccagnini A, Del Rosso M, Peschon J, Dello Sbarba P. TNF-alpha-converting enzyme cleaves the macrophage colony-stimulating factor receptor in macrophages undergoing activation. J Immunol. 2001; 166: 1583–9
- Laaksonen R, Katajamaa M, Paiva H, Sysi-Aho M, Saarinen L, Junni P, et al. A systems biology strategy reveals biological pathways and plasma biomarker candidates for potentially toxic statin-induced changes in muscle. PLoS ONE. 2006; 1: e97
- Huovila AP, Eder AM, Fuller SD. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J Cell Biol. 1992; 118: 1305–20
- Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991; 39: 741–8
- Izumi Y, Hirata M, Hasuwa H, Iwamoto R, Umata T, Miyado K, et al. A metalloprotease-disintegrin, MDC9/meltrin-gamma/ADAM9 and PKCdelta are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 1998; 17: 7260–72
- Miyamoto S, Yagi H, Yotsumoto F, Kawarabayashi T, Mekada E. Heparin-binding epidermal growth factor-like growth factor as a novel targeting molecule for cancer therapy. Cancer Sci. 2006; 97: 341–7
- Nath D, Slocombe PM, Stephens PE, Warn A, Hutchinson GR, Yamada KM, et al. Interaction of metargidin (ADAM-15) with alphavbeta3 and alpha5beta1 integrins on different haemopoietic cells. J Cell Sci. 1999; 112(Pt 4)579–87
- Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001; 52: 372–86
- Mosnier JF, Jarry A, Bou-Hanna C, Denis MG, Merlin D, Laboisse CL. ADAM15 upregulation and interaction with multiple binding partners in inflammatory bowel disease. Lab Invest. 2006; 86: 1064–73
- Maury CP, Teppo AM. Circulating tumour necrosis factor-alpha (cachectin) in myocardial infarction. J Intern Med. 1989; 225: 333–6
- Heinisch RH, Zanetti CR, Comin F, Fernandes JL, Ramires JA, Serrano CV, Jr. Serial changes in plasma levels of cytokines in patients with coronary artery disease. Vasc Health Risk Manag. 2005; 1: 245–50
- Barath P, Fishbein MC, Cao J, Berenson J, Helfant RH, Forrester JS. Detection and localization of tumor necrosis factor in human atheroma. Am J Cardiol. 1990; 65: 297–302
- Moss ML, Bartsch JW. Therapeutic benefits from targeting of ADAM family members. Biochemistry. 2004; 43: 7227–35