Abstract
ICGN/Oa mice are used to study the pathophysiological mechanisms underlying proteinuria-induced chronic kidney disease (CKD). Recently, a mutation of tensin2 gene (Tns2) was suggested to be responsible for proteinuria in the inbred ICGN mice. We identified the wild-type (+/+), heterozygous (+/nep), and homozygous (nep/nep) ICGN/Oa mice by PCR assay. The homozygotes developed proteinuria, resulting in nephrotic syndrome (NS) as early as 5 weeks and CKD by 15 weeks. However, the heterozygotes did not show the symptoms of these renal failures. These results indicate that the homozygous tensin2 mutation is necessary for the ICGN/Oa mice to develop proteinuria-induced CKD. Furthermore, we examined the time course of tubulointerstitial fibrosis and the kinetics of tubular epithelial cells (TECs) in the ICGN/Oa mice using immunohistochemical and TUNEL assays. In the renal parenchyma of the five-week-old homozygotes, the expression of α-SMA and type I collagen were higher than those in the age-matched wild-type. Additionally, increased TEC proliferation was found at 5 weeks, and increased TEC apoptosis was by 15 weeks in the homozygotes. Tubulointerstitial fibrosis precedes TEC apoptosis in the proteinuria-induced CKD model mice, and that tubulointerstitial fibrosis may be the triggering event of the disease.
INTRODUCTION
The progression of chronic kidney disease (CKD) is an increasingly common condition, often leading to complete destruction of functional kidney tissue and dependency of affected individuals on life-long treatment with dialysis or renal allograft transplantation.Citation[1] The population undergoing dialysis at the end of 2005 was about 257,000, an increase of 9,600 patients (3.87%) since 2004 in Japan.Citation[2] The escalation of CKD to an urgent matter of public health concern is a consequence of the current and anticipated burden.Citation[3] Although traditionally considered to be a marker of renal injury, there is increasing evidence that proteinuria plays a key role in the tubulointerstitial changes that ultimately lead to renal insufficiency.Citation[4],Citation[5] Both clinical and experimental data suggest that proteinuria is closely involved in pro-inflammatory and pro-fibrotic changes of the tubulointerstitium.Citation[6–8] Previous studies show that the deterioration of renal function is largely determined by the extent of tubulointerstitial alterations in many forms of renal disease, both in experimental models and in clinical patients.Citation[9],Citation[10] Tubulointerstitial fibrosis (TIF) is often recognized as an endpoint outcome of a wide range of CKD regardless of the underlying pathogenesis.Citation[9] A striking feature of this fibrosis is tubulointerstitial myofibroblast activation, which is thought to be central in the pathogenesis of renal disease.Citation[10],Citation[11] Additionally, epithelial cell loss characterized as tubular atrophy is considered to be a hallmark in the development of TIF.Citation[12],Citation[13] This pathological process not only directly contributes to the progressive loss of renal function directly, but also exacerbates the accumulation and deposition of extracellular matrix (ECM) components leading to TIF, probably because of the collapse and concentration of ECM surrounding the lost cells. In this regard, tubular atrophy and TIF are often interdependent, mutually stimulating events that ultimately lead to end stage renal failure.Citation[14]
The ICR strain-derived glomerulonephritis (ICGN) mice are the unique model strain of intractable nephrotic syndrome.Citation[15] Nephrotic syndrome (NS) was evident in the homozygotes (nep/nep) and that the renal dysfunction began to increase from 14 weeks after birth by comparing the heterozygous (+/nep) mice.Citation[15] We recently demonstrated that the tensin2 gene in the ICGN mice possessed an 8 bp nucleotide deletion, and proposed this mutation was responsible for nephrotic syndrome (NS), identified by heavy proteinuria and subsequent hypoproteinemia, hypercholesterolemia, and massive edema.Citation[16] Tensin2 is expressed in podocytes and in tubular epithelial cells.Citation[16] Tensin2 is a focal adhesion molecule and promotes cell migration on fibronectin.Citation[17] These indicate that tensin2 plays a role in regulating cell motility in kidney.
In the present study, we have identified the wild-type (+/+), heterozygous, and homozygous ICGN/Oa mice by PCR analysis, and investigated the effects of a mutation of the tensin2 gene mutation on the renal lesions among three genotypes of the inbred ICGN/Oa mice to find whether the heterozygotes show the symptoms of proteinuria-induced CKD. Additionally, we have determined the time course of TIF and tubular injuries in the homozygotes by comparing with those in the wild-types to study the pathophysiological sequence of events in the spontaneous proteinuria-induced CKD model.
MATERIALS AND METHODS
Animals and Sampling
SPF ICGN/Oa mice were kept at 21 ± 2°C and 40 ± 5 % humidity under a light/dark regimen of 12h on and 12h off, and fed a commercial laboratory diet (MF; Oriental Yeast, Tokyo, Japan) and reverse osmotic (RO) water ad libitum. The heterozygotes were prepared by mating between the homozygous males and heterozygous females.Citation[18–20] The homozygotes were prepared by mating the homozygotes. The wild-types were generated by mating the heterozygotes, and then prepared by mating the wild-types. To determine the natural course of CKD, three strains of mice were subjected to autopsy at the ages of 5, 15, and 25 weeks. Each scheduled autopsy series included five males and five females. At this time, blood samples were obtained by cardiac puncture under anesthesia (pentobarbital sodium, 50 mg/kg, i.p.). The renal capsule was removed on the paper towel to exclude water and urine in the kidney. In addition, to determine the total amount of 24 h urinary albumin, the males were placed in a metabolic cage (Sugiyamageniriki, Tokyo, Japan).
PCR Analysis
The left kidneys removed at each autopsy were at liquid N2 and stored −80°C until use. Isolation of genomic DNA from kidneys was performed with DNeasy (QIAGEN, Hilden, Germany) according to the manufacturer's instruction. PCR was performed using AccuPrimeTM Taq DNA supermix II (Invitrogen, Carlsbad, California, USA) with 2 μl of extracted DNA in a final volume 25 μl containing 0.32 μM primers. For the detection of the tensin2 mutation, the primer used was as follows:
Forward 5′-CCC ACC CCC GAT GCT CTC TG-3′
Reverse 2 5′-CCG GGG GAT TCT GCT GTG TG-3′.
After incubation at 94°C for 1 min, PCR were performed for 35 cycles at 94°C for 30 sec, 68°C for 30 sec, and 74°C for 10 sec, followed by 74°C for 1 min.
Detection of Albuminuria
Urine was collected and analyzed using SDS-PAGE. Gels were stained with SimplyBlue Safestain (Invitrogen).
Serum Analysis
The urinary and serum albumin concentrations were determined by a bromo-cresol green method using an ALB-G test kit (Denka Seiken, Tokyo, Japan). The serum total cholesterol (sTcho), BUN, and serum creatinine (sCre) concentrations were examined as described previously.Citation[15],Citation[21]
Morphometric Analysis of Tubular Lesions
The left kidney removed at each necropsy was fixed in 10% neutral-buffered formalin (pH 7.4). The transversally trimmed kidney tissues were submitted to a routine process for paraffin embedding. The sections were cut in 4 μm slices, dewaxed, and then stained with H&E and PAS. Mononuclear cell (MNC) infiltrates around renal arteries were determined on H&E-stained sections and graded, as described previously.Citation[16] Tubular injuries, characterized by the tubular dilation, epithelial cellular atrophy, and luminal cast formation were determined on PAS-stained sections and graded, as described previously.Citation[22],Citation[23] MNC infiltrations and tubular injuries were graded by an observer who was unaware of the origin of the slides.
Immunohistochemical Analyses
The right kidney removed at each autopsy was in cold 70% ethanol for 12–24 hours. The sections were prepared as described above. The methods to identify α-SMA, type I collagen, and PCNA have been described previously.Citation[24] The matrix score for α−SMA and type I collagen in the cortical interstitium were determined as described previously.Citation[24],Citation[25] Twenty random, non-overlapping fields of sections stained for PCNA were scanned, and the number of labeled TECs was counted in each fields.Citation[26]
Immunohistochemical Analysis to Identify Apoptotic Tubular Epithelial Cells
The left kidney removed at each scheduled autopsy was in cold 4% paraformaldehyde for 24–48 hours. The sections were prepared as described above. To detect apoptotic cells, a commercial kit (Apoptag, Chemicon, Temecula, California, USA) was used according to the manufacturer's instructions. Thirty to forty random, non-overlapping fields of sections stained for TUNEL were scanned, and the number of labeled TECs was counted in each field.Citation[26]
Statistical Analyses
All data were expressed as mean ± SD. For comparisons of groups, statistical analysis used was the unpaired t‐test, with the level of significance set at p < 0.05.
RESULTS
PCR Analysis
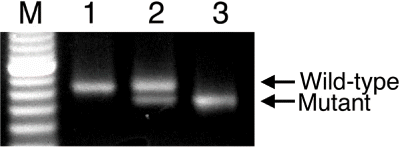
To identify the wild-type, heterozygote, and homozygote, we designed a PCR analysis using the specific primers amplifying the region, including an 8 bp nucleotide deletion in exon 18 of the tensin2 gene (Tns2). Thus, we detected an 83 bp PCR product from the wild-type allele, and a 75 bp product from the mutant. As shown in , although the single band was found from the wild-type and homozygote, respectively, the double bands were found from the heterozygotes. Thus, we developed the method for genotyping using the tensin2 mutation in the ICGN/Oa mouse.
Figure 1. PCR analysis of the ICGN/Oa mice for the tensin2 locus. The DNA extracted from the wild-type (lane 1), heterozygous (lane 2), and homozygous (lane 3) mice were genotyped using a single pair of specific primers for the wild-type and mutant alleles, respectively. Lane M was loaded with the size marker.
Body and Kidney Weights
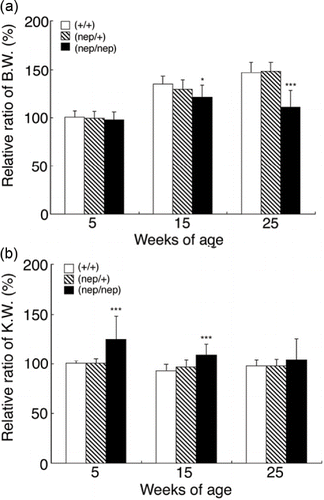
To investigate whether three strains of the ICGN/Oa mice had the physiological changes, we measured the body and kidney weights at 5, 15, and 25 weeks of age. The ICGN/Oa male mice showed higher body weight than the females (data not shown). The measured body and kidney weights were represented as the relative ratio to the average level of the five-week-old wild-types to integrate those in the males and females, respectively. As shown in , body weights of the homozygotes were lower than those of the wild-type and heterozygotes by 15 weeks and significantly lower by 25 weeks. As shown in , the kidney weights of the homozygotes were significantly higher than those of the wild-types and heterozygotes at five weeks of age. On the other hand, body and kidney weights of the heterozygotes were identical to those of the wild-type. These results show that the physiological differences could be detected in the homozygotes compared with the wild-types and heterozygotes.
Figure 2. Effects of a tensin2 mutation on body and kidney weights of the heterozygous and homozygous mice at 5, 15, and 25 weeks of age compared with the wild-types. (A) body and (B) kidney weights. These parameters are expressed as mean ± SD. *p < 0.05, ***p < 0.001 as compared with the wild-types.
Urinary and Serum Biochemistry
To investigate proteinuria, NS, and CKD in three strains of the ICGN/Oa mice, we performed biochemical analyses of urine and serum. The urine samples collected from three strains of ICGN/Oa mice were analyzed with SDS-PAGE to estimate the albumin fraction. It was found that the protein fraction with 66.1 kDa was albumin fraction. As shown in , the albumin band was identified in the homozygote but not the heterozygotes and wild-types. Furthermore, the homozygotes excreted the high concentrations of urinary albumin compared with the wild-types and heterozygotes at 5, 15, and 25 weeks (see ). Additionally, the total urinary albumin secretion in 24 hours was statistically larger than in the homozygotes. To investigate the nephrotic states in the ICGN/Oa mice, we measured serum albumin and serum total cholesterol. As shown in , the serum albumin concentrations in the homozygotes were lower than those in the wild-types and heterozygotes as early as 5 weeks and continued further until 25 weeks. As reported previously,Citation[20] the mean concentrations of serum total cholesterol in males were also higher than those in females in the ICGN/Oa mice (data not shown). We determined the serum total cholesterol concentrations in the males and females, and presented as the relative ratio to the mean concentration of the 5-week-old wild-type. As shown in , the serum total cholesterol levels were higher in the homozygotes than those in the wild-types and heterozygotes as early as 5 weeks and increased thereafter with age. The serum total cholesterol levels in the 25-week-old homozygotes were on average two-fold higher than those in the wild-types. To assess the renal dysfunction in the ICGN/Oa mice, we measured the concentrations of blood urea nitrogen (BUN) and serum creatinine. As shown in , BUN and serum creatinine levels in the homozygotes were similar between the wild-types and heterozygotes at 5 weeks, but the levels increased with age. Namely, BUN and serum creatinine levels at 25 weeks of age were 5.5- and 2.6-fold higher than in the age-matched wild-type mice, respectively. On the other hand, in terms of the biochemical analyses of urine and serum, the heterozygotes were identical to the wild-types. These results showed that the homozygotes developed proteinuria, resulting in nephrotic syndrome (NS) as early as 5 weeks and CKD by 15 weeks.
Figure 3. SDS-PAGE analysis of urine from the three strains of ICGN/Oa mice at 5, 15, and 25 weeks of age.
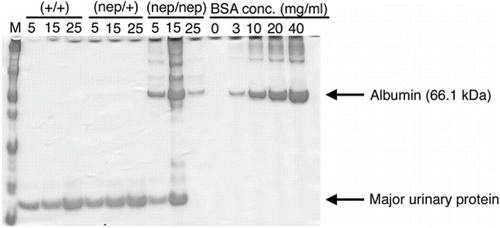
Table 1 Effects of a tensin2 mutation on urine and serum biochemical parameters in the heterozygous and homozygous mice at 5, 15, and 25 weeks of age compared with the wild-types
Histological Changes
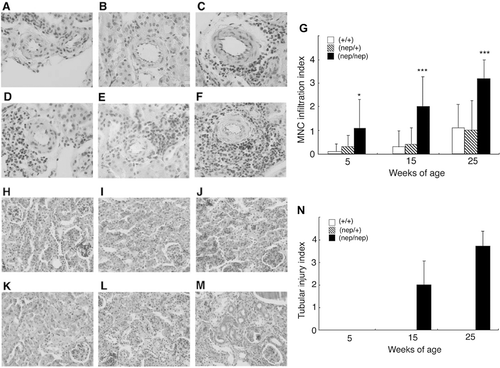
In order to investigate renal lesions in the ICGN/Oa mice, we performed histological examinations by hematoxylin and eosin (H & E) and periodic acid-Schiff (PAS) staining. Mononuclear cell (MNC) infiltrations were rarely found in the wild-type and heterozygote at 5 weeks (see and b), but detected more often by 25 weeks (see and e). Namely, MNC infiltration indexes were 0.10 ± 0.32, 0.30 ± 0.67, and 1.10 ± 0.99 in the wild-types and 0.30 ± 0.48, 0.40 ± 0.70, and 1.00 ± 1.25 in the heterozygotes at 5, 15, and 25 weeks, respectively (see ). In the homozygotes, on the other hand, MNC infiltrations were detected as early as 5 weeks (see ), and at higher frequencies with age (see ). Namely, the indices in the homozygotes were 1.10 ± 1.20, 2.00 ± 1.26, and 3.20 ± 0.79 at 5, 15, and 25 weeks, respectively. Tubular lesions were not observed in the wild-type (see and k) or heterozygotes (see and l). As shown in , the tubular structures were well preserved in the 5‐week-old homozygotes. However, the cortical area in the 15-week-old homozygotes showed the tubular dilation but also epithelial cell atrophy and luminal cast formation. These tubular lesions in the homozygotes were more evident at 25 weeks of age (see ). As shown in , the tubular injury indexes in the homozygotes were 2.00 ± 1.05 and 3.70 ± 0.56 at 15 and 25 weeks of age, respectively. These results indicate induction of MNC infiltration was detected at 5 weeks of age, and tubular injuries became evident by 15 weeks in the homozygotes.
Figure 4. Histopathology of the renal cortex in the ICGN/Oa mice. Representative photographs of renal arteries at 5 weeks of the (A) wild-types, (B) heterozygotes, and (C) homozygotes, and at 25 weeks of the (D) wild-types, (E) heterozygotes, and (F) homozygotes. (G) Semiquantification of these histological findings. Mononuclear cell infiltration indexes are expressed as mean ± SD. *p < 0.05, ***p < 0.001 as compared with the wild-types. Representative photomicrographs of renal tubules at 5 weeks of the (H) wild-types, (I) heterozygotes, and (J) homozygotes, and at 25 weeks of the (K) wild-types, (L) heterozygotes, and (M) homozygotes. (N) Semiquantification of these histological findings. Tubular injury indexes are expressed as mean ± SD.
Tubulointerstitial Fibrosis
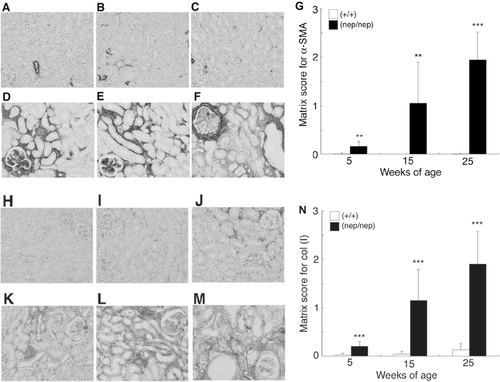
To determine the numbers of myofibroblasts in the tubulointerstitial space, we performed an immunohistochemical analysis of the tubulointerstitium using specific antibodies for α-SMA. In the renal cortex of the wild-types, immunostaining of the α-SMA antigen was almost localized to normal renal vessels (see –). However, in the homozygotes, marked expression of α-SMA was found as early as 5 weeks, and it increased with the age (see –). As shown in , the matrix scores for α‐SMA in the homozygotes were 0.16 ± 0.10, 1.05 ± 0.64, and 1.94 ± 0.58 at 5, 15, and 25 weeks of age, respectively. To examine the expression of ECM in the tubulointerstitial spaces, we also performed immunohistochemical analysis using the antibody for type I collagen. The expression was low in the renal cortex of the wild-type (see -), whereas in the homozygotes, the expression was found abundantly as early as 5 weeks, and increased with age (see –). As shown in , the matrix scores for type I collagen in the homozygotes were 0.21 ± 0.10, 1.15 ± 0.64, and 1.90 ± 0.67 at 5, 15, and 25 weeks, respectively. These results show that tubulointerstitial fibrosis was evident as early as 5 weeks in the homozygotes.
Figure 5. Time courses of the expression of α-SMA and type I collagen in the ICGN/Oa mice. Representative photomicrographs of α-SMA immunostain at 5 weeks of the (A) wild-types and (B) homozygotes, 15 weeks of the (C) wild-types and (D) homozygotes, and 25 weeks of the (E) wild-types and (F) homozygotes. (G) Semiquantification of these histological findings. Matrix scores for α‐SMA are expressed as mean ± SD. Representative photomicrographs of type I collagen immunostain at 5 weeks of the (H) wild-types and (I) homozygotes, 15 weeks of the (J) wild-types and (K) homozygotes, and 25 weeks of the (L) wild-types and (M) homozygotes. (N) Semiquantification of these histological findings. Matrix scores for type I collagen are expressed as mean ± SD. **p < 0.01, ***p < 0.001 as compared with the wild-types.
Proliferating and Apoptotic TECs
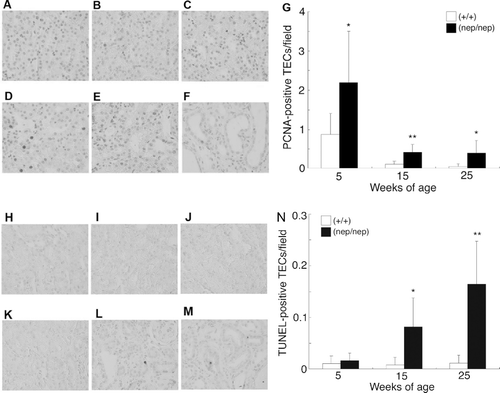
To quantify the proliferating TEC in the homozygotes, we performed immunohistochemical analysis using the antibody against PCNA. In the wild-types, PCNA-positive TECs were found at 5 weeks (see ), but few were detected at 15 and 25 weeks (see and c). Namely, the proliferating TEC scores in the wild-types were 0.87 ± 0.53, 0.11 ± 0.08, and 0.05 ± 0.07 at 5, 15, and 25 weeks, respectively (see ). In the homozygotes, the numbers of proliferating TECs at 5 weeks were also higher than those at 15 and 25 weeks. However, their proliferating TEC were higher than those in the age-matched wild-types (see –). Namely, the proliferating TEC scores in the homozygotes were 2.19 ± 1.30, 0.41 ± 0.21, and 0.39 ± 0.33 at 5, 15, and 25 weeks, respectively (see ). We then determined the number of the apoptotic TECs by TUNEL assay. Few TUNEL-positive TECs were found in the wild-type (see –). The homozygotes at 5 weeks, the numbers of TUNEL-positive TEC showed the same level as those in the wild-types (see ). At 15 and 25 weeks however, TUNEL-positive TECs were significantly increased (see –). As shown in , the TUNEL-positive TEC scores in the homozygotes were 0.016 ± 0.015, 0.088 ± 0.054, and 0.164 ± 0.086 at 5, 15, and 25 weeks, respectively. These results indicate that TECs transiently proliferate at 5 weeks of age and disappear by apoptosis with age in the homozygotes.
Figure 6. Time courses of the proliferating and apoptotic TECs in the ICGN/Oa mice. Representative photomicrographs of PCNA immunostain at 5 weeks of the (A) wild-types and (B) homozygotes, 15 weeks of the (C) wild-types and (D) homozygotes, and 25 weeks of the (E) wild-types and (F) homozygotes. (G) Semiquantification of these histological findings. The numbers of PCNA-positive TECs are expressed as mean ± SD. Representative photomicrographs of TUNEL at 5 weeks of the (H) wild-types and (I) homozygotes, 15 weeks of the (J) wild-types and (K) homozygotes, and 25 weeks of the (L) wild-types and (M) homozygotes. (N) Semiquantification of these histological findings. The numbers of TUNEL-positive TECs are expressed as mean ± SD. *p < 0.05, **p < 0.01 as compared with the wild-types.
DISCUSSION
The ICGN/Oa is an inbred strain of mice derived from an outbred Yok:ICR colony at the National Institute of Health (Japan) in 1986 and develops nephrotic syndrome accompanying glomerular lesion.Citation[16] Electron microscopic studies showed abnormal architecture of the glomerular basement membrane (GBM) and loss of the epithelial cell foot process that took place from the maturing glomerular stage onward in the ICGN mice.Citation[27],Citation[28] The ICGN homozygous mice showed urinary excretion of albumin during the suckling stage.Citation[21] The homozygotes were used as an appropriate control for pathophysiological studies on human CKD.Citation[22],Citation[24] We identified a mutation in tensin2 as a candidate gene responsible for proteinuria in the ICGN mice.Citation[16] Furthermore, we have taken advantage of the tensin2 mutation, 8 bp deletion, and devised the PCR method, which makes use of one primer pair, simultaneously to identify the two distinct genotypes, the wild-type and mutant alleles. Accordingly, we could detect the tensin2 mutation in the ICGN/Oa mice, and genetically identified the wild-types, heterozygotes, and homozygotes, respectively. We examined the renal lesions among the three genotypes of ICGN/Oa mice at 5, 15, and 25 weeks of age using physiological, biochemical, and histological methods. In the present study, we found NS and MNC infiltration at 5 weeks, tubular injuries and renal dysfunction by 15 weeks, and weight loss by 25 weeks in the homozygotes. Regarding MNC infiltration, the interstitial inflammatory cells include macrophage marker and Mac1-positive cells (data not shown). On the other hand, the heterozygotes were identical to the wild-types in these renal lesion parameters. Furthermore, we prepared the heterozygotes by mating between the wild-types and homozygotes. By the urine and serum biochemical analysis, we could not find any signs of proteinuria, NS, or CKD in the heterozygotes that were generated by backcrossing the homozygotes with the wild-types (data not shown). Thus, the heterozygotes did not show symptoms of proteinuria, NS, and CKD. In the ICGN mice, eight nucleotides are deleted from exon 18 of the tensin2 gene, resulting in a frameshift in the mRNA and producing a truncation mutation.Citation[16] By northern and western blot analyses, the expression of tensin2 was lost in the ICGN mice.Citation[16] Accordingly, it is likely that the homozygotes lose expression of tensin2 in the kidneys and the function of the glomerular filtration barrier, which leads to proteinuria. On the other hand, the heterozygotes express tensin2 gene sufficiently and therefore kept the -filtration barrier function.
Here, in Osaka University, we have been the inbreeding with sister-brother mating system for 40 generations, and established an inbred strain as ICGN/Oa. In the present study, we have demonstrated that the homozygous mutations of the tensin2 gene are necessary for the ICGN/Oa mice to develop proteinuria-induced CKD. However, the individual level of albuminuria in (ICGN × MSM) F1 × ICGN backcross progenies has not been reported to be segregated into two groups.Citation[16] Therefore, it was proposed that the nephrotic syndrome of the ICGN mice is controlled by multigenes.Citation[16] Although the various albumin concentrations in the urine samples were found in the ICGN/Oa homozygotes, they varied according to the sampling time partly (data not shown). In the present study, we found that the total amounts of 24 hour urinary albumin of the homozygotes were statistically different from those of both the wild-types and heterozygotes. Additionally, in the QTL analysis, we detected only one significant QTL located in the telomeric region of Chr 15 with a very high LOD score.Citation[16] Whether proteinuria is modified by unknown factors or not awaits further investigation, though it is therefore unlikely that the ICGN/Oa mice develop proteinuria-induced CKD by other genetic factors.
It was reported that TIF develops regardless of the initiating insult and the extent of fibrosis correlated with renal dysfunction.Citation[29] ?This process of TIF is characterized by excessive deposition of ECM components and the consequent destruction of the normal tissue architecture. We found that the numbers of myofibroblasts in the homozygotes were significantly higher than those in the wild-types as early as 5 weeks. Furthermore, changes of the myofibroblast number paralleled those of the components of the type I collagen. However, tubular injuries in the homozygotes, characterized by tubular atrophy, tubular dilation, and luminal cast formation, became more marked than in the wild-types by 15 weeks. The TEC apoptosis is pathologically related to the tubular atrophy and renal tissue loss that occur in animals,Citation[30] as well as in humansCitation[31] with chronic obstruction nephropathy. The numbers of apoptotic TECs in the homozygotes increased in the homozygotes by 15 weeks, although they were not much different from those in the wild-types by 5 weeks. We found that TIF parallel TEC apoptosis in the ICGN/Oa mice. Although it is possible that TEC apoptosis may begin at earlier age, TIF may affect TEC apoptosis and tubular injuries in proteinuria-induced CKD. Eddy described that as ECM continues to accumulate and expand within the interstitial space, they begin to show destructive effects on the kidney structure and subsequently function.Citation[14] Furthermore, the renal tubules, which account for 80% of the total kidney volume, ultimately become victims of the destructive fibrogenic process that in many situations they have helped to initiate.Citation[14] Not only does the fibrotic interstitial matrix isolate tubules for their oxygen supply, but the postglomerular peritubular capillaries themselves are obligated through destruction by fibrosis.Citation[14] Studies in vitro show that tubular cell death occurs via caspase-dependent apoptosis associated with increased oxidative stress.Citation[32] The apoptosis promote inflammatory response and result in the matrix synthesis and deposition by native renal cells.Citation[31] Additionally, multiple studies have analyzed the relationship among ECM and cell morphology, proliferation, and apoptosis.Citation[30],Citation[31] Accordingly, the causal relationship between TIF to TEC apoptosis and tubular injuries remains to be investigated further.
Kidneys of the homozygotes were heavier than those of the wild-type and heterozygotes at 5 weeks. As described above, about 80% of total kidney volume is composed of TEC.Citation[14] Klahr described that TEC proliferation was paralleled with the dry weight of the chronic obstructive uropathy (COU) kidney.Citation[31] In the experiments shown in and a, the numbers of proliferating TEC paralleled the kidney weight of the 5-week-old homozygotes. Consistently the TEC proliferation indexes significantly were higher after 5 weeks in the ICGN/Oa mice, and the TEC apoptosis increased by 15 weeks. Truong et al. reported that TEC proliferation peak immediately preceded the surge in TEC apoptosis in the COU.Citation[30] In the homozygotes at 5 weeks, TEC proliferation was induced tentatively and TEC apoptosis was not yet activated for immunostaining analysis.
In conclusion, three strains of the ICGN/Oa mice could be identified by the tensin2 mutation using PCR analysis. The homozygotes showed proteinuria, resulting in NS and CKD. However, the heterozygotes showed no symptoms of these renal failures. Additionally, TIF develops at the early age, and tubular injuries including TEC apoptosis develops later in the spontaneous proteinuria-induced CKD model mice.
DECLARATION OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Acknowledgments
The authors would like to thank Yasuhiko Kozawa, Katsutoshi Terasaka, and Takashi Ito for their technical assistance.
REFERENCES
- Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002; 13: 2600–2610
- Nakai S, Masakane I, Akiba T, et al. Overview of regular dialysis treatment in Japan (as of Dec. 31, 2005). Ther Apher Dial. 2007; 11: 411–441
- Owen WF, Jr. Patterns of care for patients with chronic kidney disease in the United States: Dying for improvement. J Am Soc Nephrol. 2003; 14: S76–S80
- Benigni A, Zoja C, Remuzzi G. The renal toxicity of sustained glomerular protein traffic. Lab Invest. 1995; 73: 461–468
- D'Amico G, Bazzi C. Pathophysiology of proteinuria. Kidney Int. 2003; 63: 809–825
- Eddy AA. Interstitial nephritis induced by protein-overload proteinuria. Am J Pathol. 1989; 135: 719–733
- Ruggenenti P, Perna A, Mosconi L, Pisoni R, Remuzzi G. Urinary protein excretion rate is the best independent predictor of ESRF in non-diabetic proteinuric chronic nephropathies. Gruppo Italiano di Studi Epidemiologici in Nefrologia (GISEN). Kidney Int. 1998; 53: 1209–1216
- Burton C, Harris KP. The role of proteinuria in the progression of chronic renal failure. Am J Kidney Dis. 1996; 27: 765–775
- Eddy AA. Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol. 1996; 7: 2495–2508
- Essawy M, Soylemezoglu O, Muchaneta-Kubara EC, Shortland J, Brown CB, el Nahas AM. Myofibroblasts and the progression of diabetic nephropathy. Nephrol Dial Transplant. 1997; 12: 43–50
- Hewitson TD, Becker GJ. Interstitial myofibroblasts in IgA glomerulonephritis. Am J Nephrol. 1995; 15: 111–117
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002; 283: F861–F875
- Chevalier RL. Molecular and cellular pathophysiology of obstructive nephropathy. Pediatr Nephrol. 1999; 13: 612–619
- Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000; 15: 290–301
- Mizuno S, Yue BF, Okamoto M, Horikawa Y, Kurosawa T. Diffuse glomerulosclerosis without tubular injury does not directly manifest renal dysfunction in nephrotic mice (ICGN strain). Exp Nephrol. 1997; 5: 498–507
- Cho A, Uchio-Kozue K, Torigai T, et al. Deficiency of the tensin2 gene in the ICGN mouse: An animal model for congenital nephritic syndrome. Mamm Genome. 2006; 17: 406–416
- Chen H, Duncan IC, Bozorgchami H, Lo SH. Tensin1 and a previously undocumented family member, tensin2, positively regulate cell migration. Proc Natl Acad Sci USA. 2002; 99: 733–738
- Kurosawa T, Okamoto M, Yamada K, Yue BF. Nephrosis (nep): A new mouse mutation which causes albuminuria and other symptoms of nephrosis. Mouse Genome. 1993; 91: 876–878
- Mizuno S, Mizuno-Horikawa Y, Kurosawa T. Immunohistochemical analysis of molecular events in tubulo-interstitial fibrosis in a mouse model of diffuse mesangial sclerosis (ICGN strain). J Vet Med Sci. 2001; 63: 299–307
- Mizuno-Horikawa Y, Mizuno S, Tamura S, Kurosawa T. Advanced glomerulosclerosis is reversible in nephrotic mice. Biochem Biophys Res Commun. 2001; 284: 707–713
- Mizuno S, Mizuno-Horikawa Y, Yue BF, Okamoto M, Kurosawa T. Nephrotic mice (ICGN strain): A model of diffuse mesangial sclerosis in infantile nephrotic syndrome. Am J Nephrol. 1999; 19: 73–82
- Mizuno S, Kurosawa T, Matsumoto K, Mizuno-Horikawa Y, Okamoto M, Nakamura T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J Clin Invest. 1998; 101: 1827–1834
- Kliem V, Johnson RJ, Alpers CE, et al. Mechanisms involved in the pathogenesis of tubulointerstitial fibrosis in 5/6-nephrectomized rats. Kidney Int. 1996; 49: 666–678
- Mizuno S, Matsumoto K, Kurosawa T, Mizuno-Horikawa Y, Nakamura T. Reciprocal balance of hepatocyte growth factor and transforming growth factor-beta 1 in renal fibrosis in mice. Kidney Int. 2000; 57: 937–948
- Sawashima K, Mizuno S, Mizuno-Horikawa Y, Shimada A, Kudo T, Kurosawa T. Expression of alpha-smooth muscle actin and fibronectin in tubulointerstitial lesions of cats with chronic renal failure. Am J Vet Res. 2000; 61: 1080–1086
- Chevalier RL, Goyal S, Kim A, Chang AY, Landau D, LeRoith D. Renal tubulointerstitial injury from ureteral obstruction in the neonatal rat is attenuated by IGF-1. Kidney Int. 2000; 57: 882–890
- Ogura A, Asano T, Suzuki O, et al. Hereditary nephrotic syndrome with progression to renal failure in a mouse model (ICGN strain): Clinical study. Nephron. 1994; 68: 239–244
- Ogura A, Fujimura H, Asano T, et al. Early ultrastructural glomerular alterations in neonatal nephrotic mice (ICGN strain). Vet Pathol. 1995; 32: 321–323
- D'Amico G, Ferrario F, Rastaldi MP. Tubulointerstitial damage in glomerular disease: Its role in the progression of renal damage. Am J Kidney Dis. 1995; 2: 124–132
- Truong LD, Petrusevska G, Yang G, et al. Cell apoptosis and proliferation in experimental chronic obstructive uropathy. Kidney Int. 1996; 50: 200–207
- Klahr S. Obstructive nephropathy. Kidney Int. 1998; 54: 286–300
- Docherty NG, O'Sullivan OE, Healy DA, Fitzpatrick JM, Watson RW. Evidence that inhibition of tubular cell apoptosis protects against renal damage and development of fibrosis following ureteric obstruction. Am J Physiol Renal Physiol. 2006; 290: F4–F13